
分子对接(Smina/Vina/Glide)
--石禹龙
Smina (Linux)
Smina: 基于Autodock Vina的对接程序,用法和Vina十分相似
一. 配体准备
采用mgltools生成配体的pdbqt格式文件:
- conda activate mgltools
- prepare_ligand4.py -l ligand.mol2 -o ligand.pdbqt
二. 受体准备
用pdb2pqr给蛋白质质子化,生成pqr文件,再用mgltools生成受体的pdbqt文件:
- conda activate pdb2pqr
- pdb2pqr_cli --ff=amber --ffout=amber --chain --with-ph=7 receptor.pdb receptor.pqr
- conda activate mgltools
- prepare_receptor4.py -r receptor.pqr -o receptor.pdbqt -C -U nphs_lps_waters_nonstdres
三. Smina对接
对接方式一:自定义盒子位置
- smina --seed 0 --config conf.txt -r receptor.pdbqt -l ligand.pdbqt -o result.sdf
注:固定随机种子方便后续重现结果。conf.txt为包含对接盒子中心/大小、输出构象数等参数的文件,在这里举例如何只输出一个打分最好的构象,格式jis如下:center指对接盒子的中心,size指盒子的长宽高,num_modes指输出构象数,对接打分见对接构象文件。
- center_x = 23
- center_y = 10
- center_z = 1
- size_x = 24
- size_y = 18
- size_z = 22
- num_modes = 1
对接方式二:以参考配体作为对接盒子中心,autobox_add指往各维度扩展多少距离(默认为4埃),ligand-old.pdb指晶体结合口袋中已有的配体文件,用于定位坐标。
- smina --seed 0 --autobox_ligand ligand-old.pdb --autobox_add 8 -r receptor.pdbqt -l ligand.pdbqt -o result.sdf --num_modes 1
Vina (Linux)
用法和smina差不多,不重复介绍了,多看看vina --help.
Maestro-glide
各个版本的glide用法接近,通过搜索关键词即可找到对应模块,这里以2015版为例。
一. 配体准备
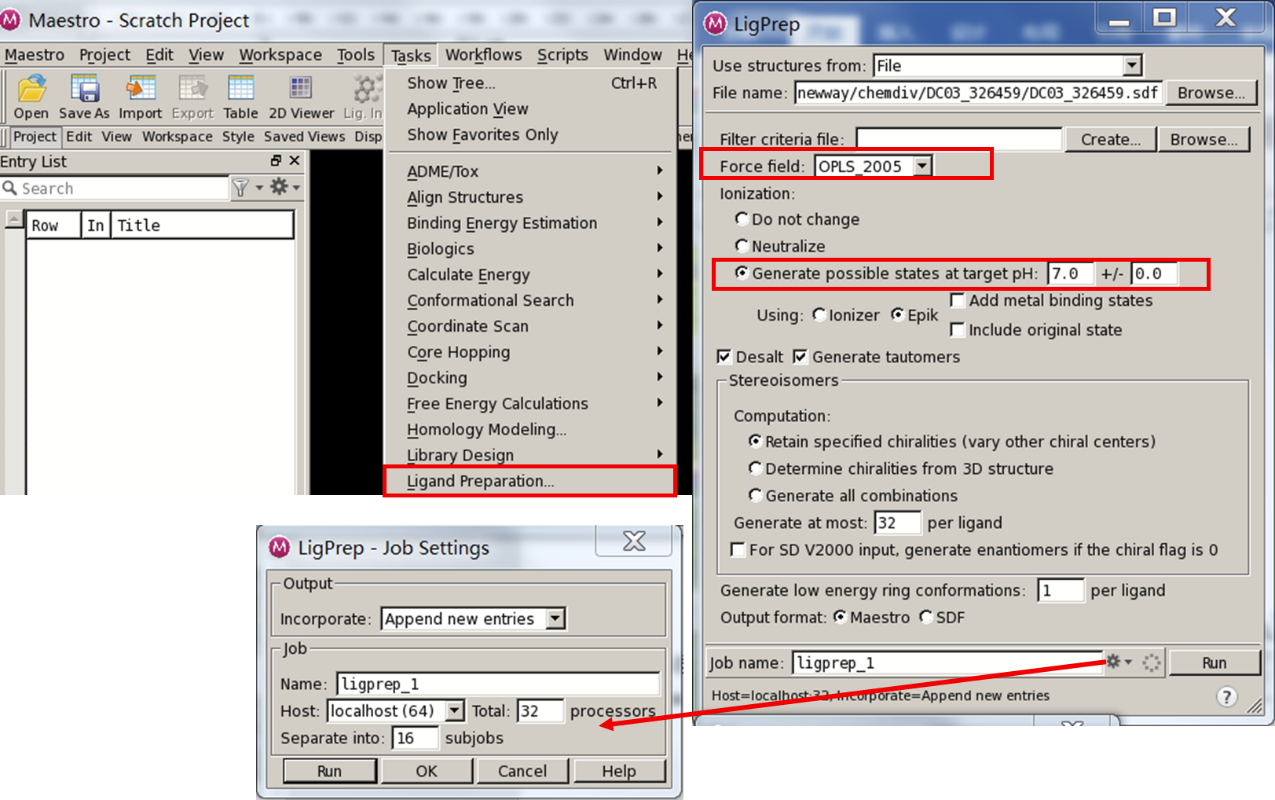
先在Maestro-Project下面更改工作目录,然后Tasks--Ligand Preparation下导入要对接的小分子整合文件,力场推荐OPLS_2005,pH根据需要调整,在job name右边设置核数和要分成多少个子任务,run。最终得到*.maegz配体文件。
二. 蛋白质准备
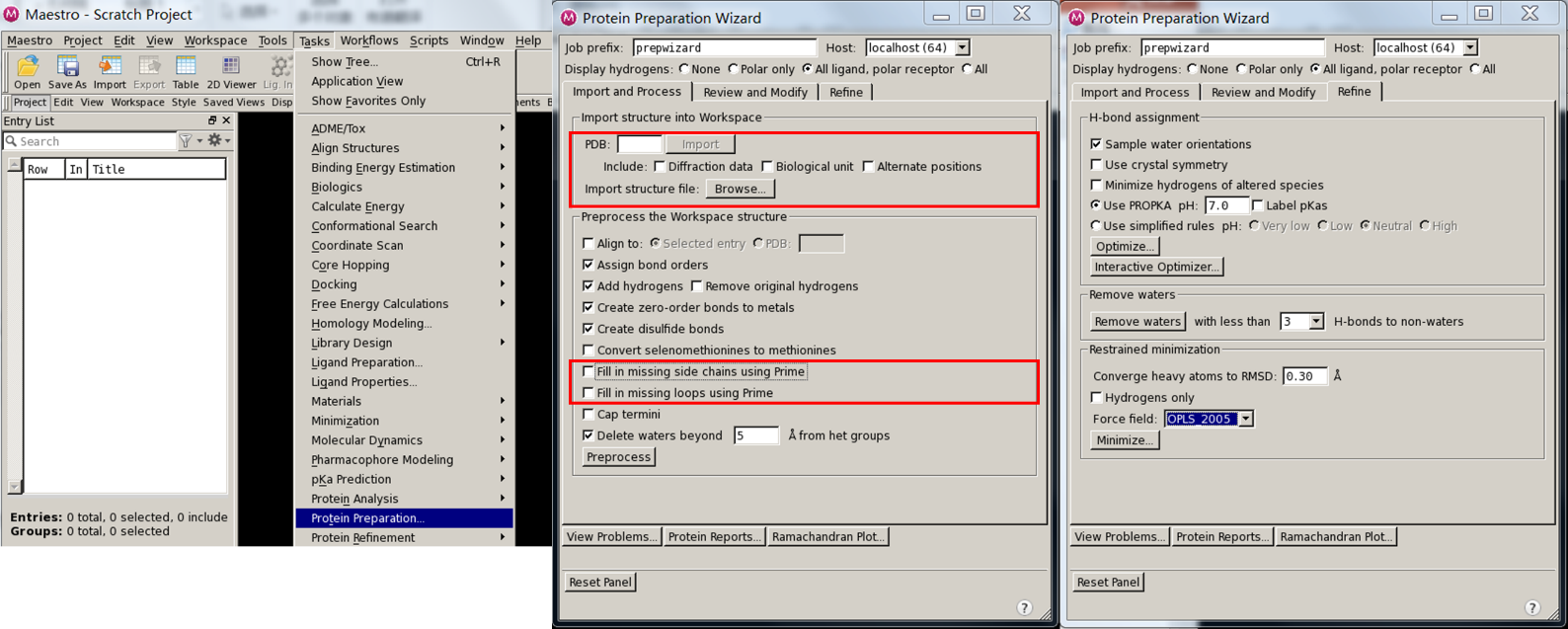
在Task—Protein Preparation Wizard下准备蛋白质文件,可以在PDB框中用PDB ID导入蛋白质,也可以用Browse导入本地的蛋白质文件,在预处理中可以选择是否补全缺少的侧链和loop(推荐用swiss model补链,然后用amber或gromacs进行动力学优化),Preprocess。完成预处理后,点击Refine进行精炼,点击Optimize,OPLS_2005力场下进行最小化优化。得到*ref.mae蛋白文件。
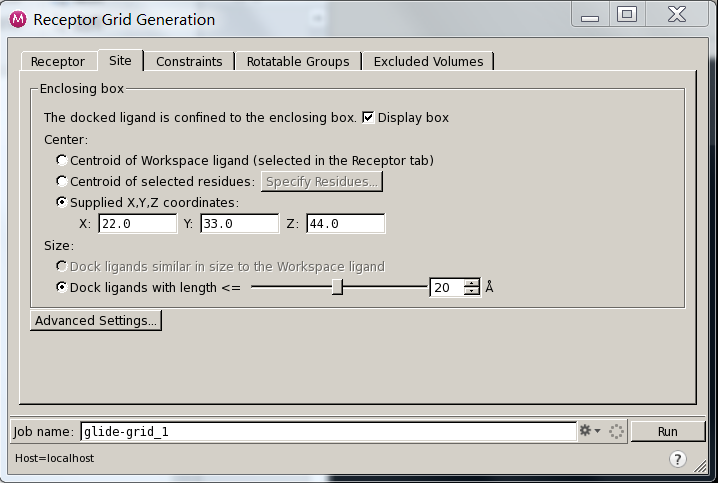
三. 格点文件准备
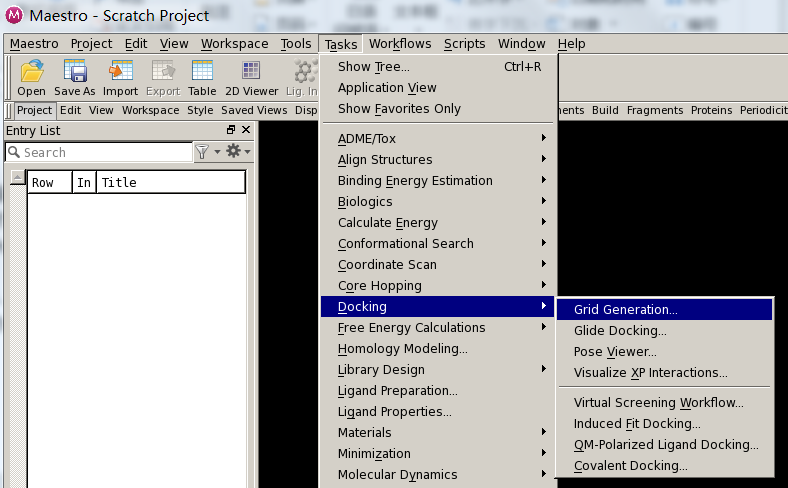
点击maestro—tasks—Docking—grid Generation生成对接盒子的格点文件。
此处有两种方式:
1. 根据蛋白中已有的配体位置作为参考,生成格点文件。
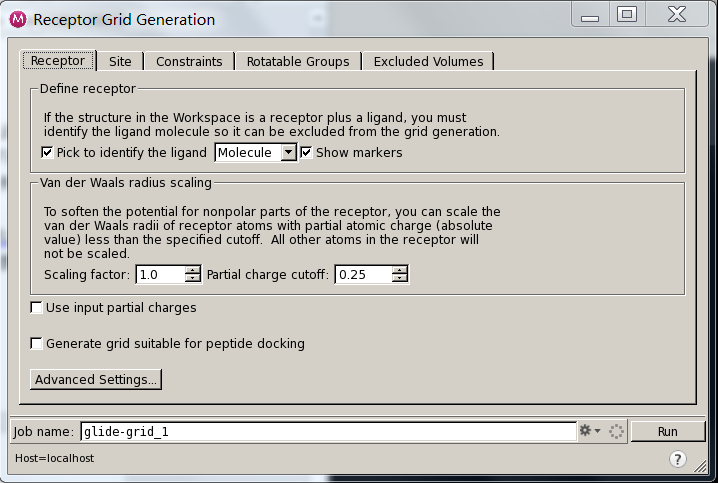
2. 自己手动设置对接位置的格点文件。这一步可以在pymol中用gridbox.py画盒子大致估摸下对接盒子的XYZ以及宽度。
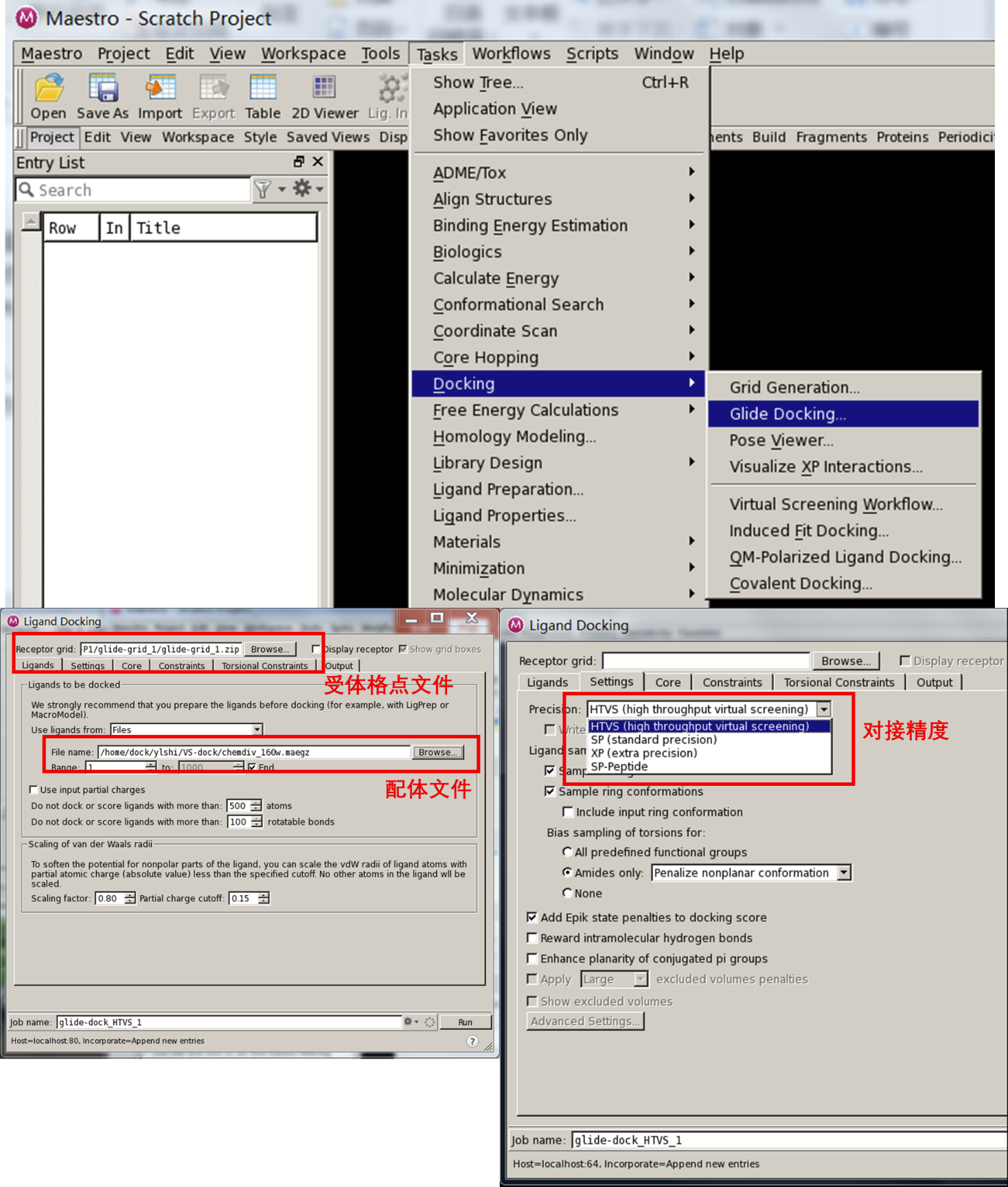
四. 分子对接/虚拟筛选
点击maestro—Tasks—Docking—Glide Docking,进行对接。上传受体的格点文件和要对接的配体文件,在Settings中设置对接精度,化合物少可以高精度,大批量时可以先HTVS初筛一遍,再高精度筛一遍。
五. 结果查看
根据对接打分、原子效率打分等评判标准选择化合物。