
固定电荷力场学习笔记
2022年1月13日 上午10:22
paper
Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview
S. Riniker
J Chem Inf Model 2018 Vol. 58 Issue 3 Pages 565-578
Accession Number: 29510041 DOI: 10.1021/acs.jcim.8b00042
在分子动力学模拟和Monte Carlo模拟中,力场可以简单理解为体系中原子的相互作用.
The interactions between the atoms in the system are defined via potential energy function of the atomic coordinates also called a force field.
经典的固定电荷力场的基础表达形式可以追述到1969年,这些年来,力场虽然变化,但是最基础的表达形式一直没有变化
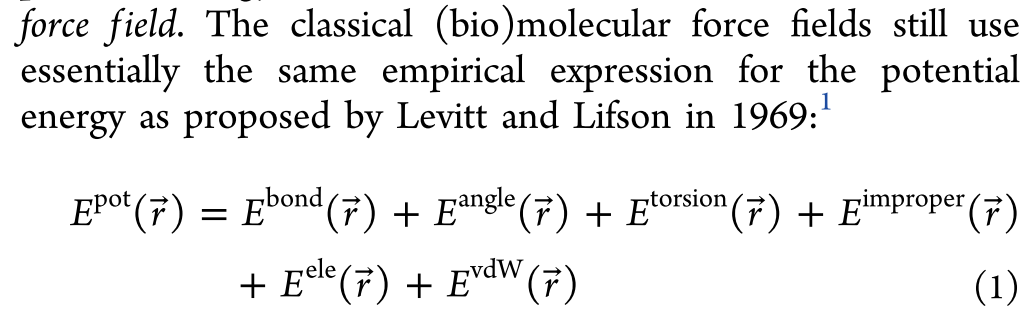
在固定电荷力场中,effective partial charges 不会随着体系构象和周围化学微环境的变化而发生改变。比起可极化力场和量子化学模型,这种简化很大程度上减少了动力学模拟的计算量。
在过去数十年的时间里,固定电荷力场被广泛地应用,并被证明能够很好地去解释化学和生物体系上的问题。
经典分子动力学模拟可以用来模拟蛋白质、核酸、脂类、糖类和有机小分子化合物(考虑规范的书面用语?)
The first, formal charge, is an integer property that is essential for the correct valence representation of a molecule. Together with atomic valences, bond order and the connectivity, this field is defines the identity of a molecule.
The second type of charge, partial charge, is a floating point property used in computational chemistry and molecular modeling. This value is used to represent the electronic distribution/wave-function of a molecule by approximating the molecule's electrostatic field with a set of point charges located at each atom.
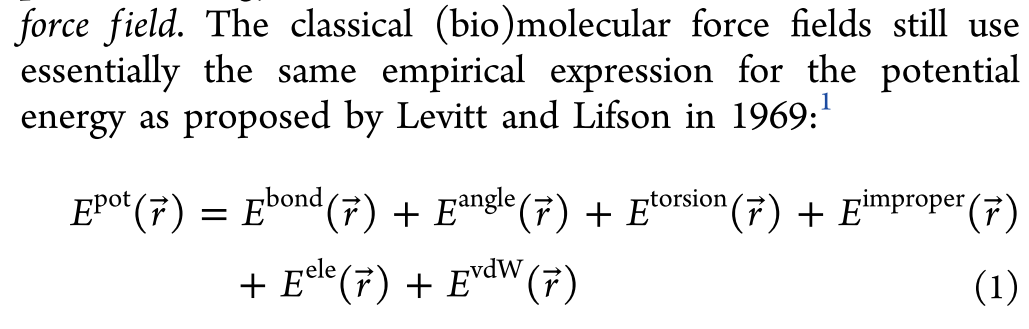
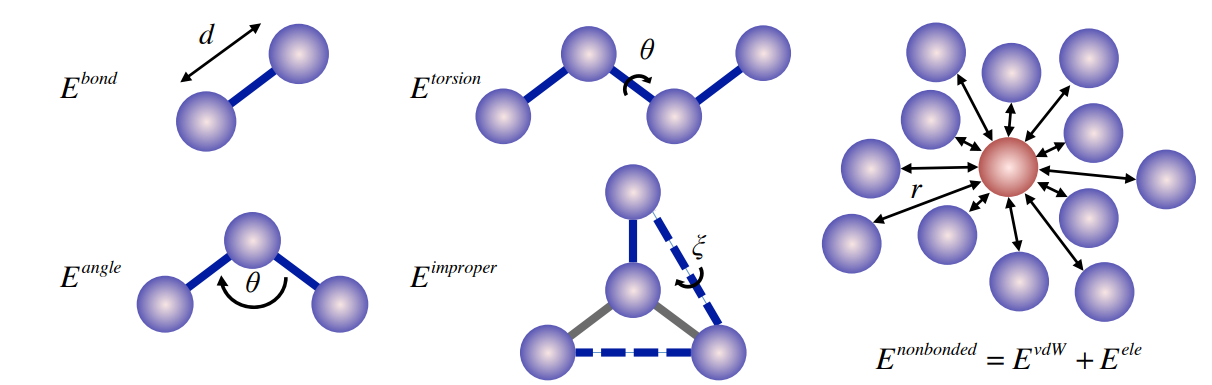
improper
inproper dihedral-angle bending ( or out-of-plane distorsions)
二面角的弯曲
目前固定电荷力场有4个主流的形式:
AMBER
CHARMM
GROMOS
OPLS
除了OPLS,另外三个力场的发展都是从氨基酸和核酸开始
OPLS力场最初是从有机溶剂开始发展的
In general, 用于有机小分子的力场,不应该去用于蛋白质和核酸分子的模拟
此外,不同的力场形式之间也是有相互影响的,比如
1)初始的OPLS力场借鉴了AMBER力场的bond terms;
2)AMBERff94力场,借鉴
可OPLS中的vdW参数;
3)CHARMM22中借鉴了OPLS中的aromatic side chains 参数。

Q: 需要注意的是OPLS力场中的有些部分是在商业化的软件当中,这里是指OPLS力场被商业化,不能公开使用吗?
Note that the OPLS force fields OPLS_2005, OPLS2.0, and OPLS3 are part of commercial software, while OPLS-AA is still actively maintained and freely distributed by the Jorgensen group. The other force-field families are publicly available.
此外,在历史上的力场发展中,很多力场在使用时需要去指定特定的水模型
bond stretching and bond angle bending
根据运动公式,想要获得准确的积分,最大的时间步长只能设置到0.5fs,
而我们通常会使用SHAKE和LINCS算法去对键进行限制(可以限制氢键,也可以限制所有的键),这样时间间隔可以增加到1-2fs
Q: 如果在对键长进行限制的时候,只限制了与H相连的键长,那么整个体系的步长调整到2fs是合理的吗?此外,这个0.5fs和2fs对应到md.mdp参数中的哪一项,是人为控制的吗?
- hard degrees of freedom中的hard如何理解?是指自由度很低的意思??
为了更好地与分子振动光谱一致,在CHARMM力场中以Urey-Bradley形式引入了额外的1-3相互作用项

需要注意的是,在实际的模拟中,除非是完全刚性的分子,不然最好不要对键角进行限制,会产生不合理的热力学和动力学性质
键伸缩项和键角弯曲项的参数化过程来源主要有3个:
1)晶体结构数据;
2)光谱实验数据;
3)量化计算数据。
Dihedral-Angle Torsion
这里主要是对1-4相互作用的处理方式的不同,主要公式表达形式采用不同级数的泰勒展开

the quality of torsional dihedral-angle terms 对蛋白质骨架和侧链特别重要
在不同的力场类型,以及同一个力场类型的不同版本中,rigidity和二级结构倾向性是不同的
目前比较流行的AMBER ff14SB力场中,对ff94和ff99SB中出现的问题进行了矫正
目前分子力场方法主要分为两大类:固定电荷力场和可极化力场
固定电荷力场
Fixed-Charge Atomistic Force Fields
Amber力场
Asissted Model Building and Energy Refinement
由Peter A.Kollman发展(找到原始文献,标注年份)
Q1: 描述固定电荷力场和可极化力场在原理上的主要区别
Q2: Amber手册上说,可极化力场对于描述分子间相互作用和重现分子间的氢键作用比addtive force field要好,具体好多少呢,有没有具体的数值体现

Q3: MMGBSA在计算的时候,氢键形成的角度和距离对其影响是多大?
固定电荷力场重现生物复合物体系中相互作用的表现如何?
可极化力场是否比固定电荷力场的重现性和和可重复性更好一些?
如果可极化力场的表现更好,能否在重要的PPI界面,用可极化力场进行优化,或者先用别的力场进行平衡,后面再用可极化力场进行优化?