
3nhe的min1构象的MMGBSA
2022年5月19日
配体小分子处理
- #22220
- antechamber -i ICU30.mol2 -fi mol2 -o lig.gjf -fo gcrt
- #修改lig.gjf
- --Link1--
- %chk=molecule
- %mem=4GB
- %nproc=8
- #B3LYP/6-31G* scrf=(solvent=water) SCF=tight Test Pop=MK
- iop(6/33=2) iop(6/42=6) opt
- nohup g09 lig.gjf &
- antechamber -i lig.log -fi gout -o lig.prep -fo prepi -c resp
- parmchk -i lig.prep -f prepi -o lig.frcmod -a y
- #最后得到lig.frcmod; lig.prep; NEWPDB.PDB
- #NEWPDB.PDB的坐标会有改变,可以对比未处理签的坐标文件修改,align一下,再修改
- #22208:以下的操作步骤可以不进行
- /home/jawang/zjfang/ligprep
- tleap
- source oldff/leaprc.ff14SB
- source leaprc.gaff
- loadamberparams lig.frcmod
- loadamberprep lig.prep
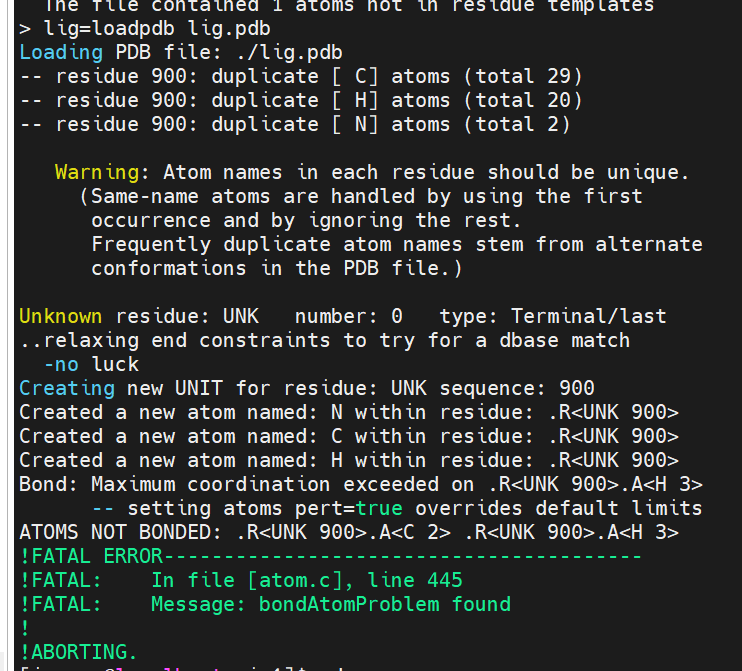
- lig=loadpdb NEWPDB.PDB
- saveamberparm lig lig.prmtop lig.inpcrd
- quit
- ambpdb -pqr -p lig.prmtop < lig.inpcrd > lig.pdb
蛋白质处理
需要先对蛋白质进行质子化预测:
- #激活pdb2pqr:
- conda activate pdb2pqr预测
- #质子化
- pdb2pqr30 min1.pdb min1.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output min1_out.pdb
- #Tleap处理蛋白质:准备好空白蛋白质.pdb和处理好的配体.pdb文件
- 空蛋白质.pdb中要包含ZN2+,需要检查与ZN2+形成配位键的几个残基的带电情况,做好记录
- #进入tleap:
- tleap
- #加载力场:
- source oldff/leaprc.ff14SB
- source leaprc.gaff
- #加载用于ZN2+的力场参数
- loadAmberParams frcmod.ions234lm_1264_tip3p
- #locate一下目录
- loadAmberParams frcmod.ionsjc_tip3p
- loadamberparams lig.frcmod
- loadamberprep lig.prep
- #加载定义蛋白质和配体
- pro=loadpdb min1_out.pdb #min1.pdb需要去掉所有氢,同时把Zn改成ZN并对齐
- lig=loadpdb NEWPDB.PDB #NEWPDB.PDB的坐标需要改,对照未处理的配体小分子的坐标修改
- #load蛋白和配体的结构之后,有二硫键的需要添加二硫键,
- bond pro.22.SG pro.96.SG
- (这条命令是指pro中第残基22的SG原子与pro中残基96的SG原子形成共价键,注意每一个二硫键都要单独用一条命令进行bond)
- #定义com文件是pro和lig的组合
- com=combine {pro lig}
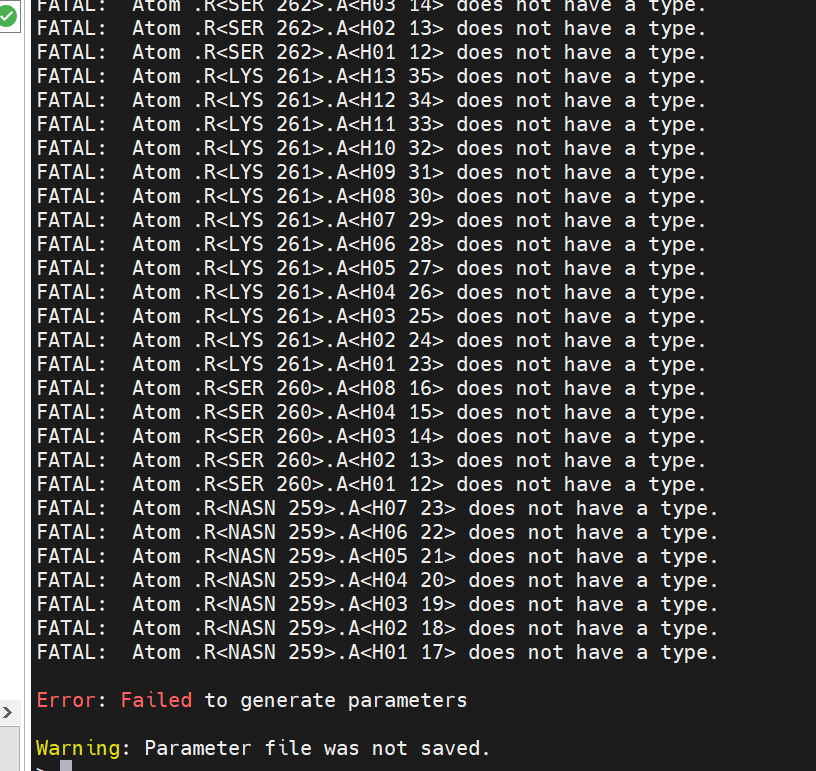
- #报错原因,可能是蛋白质文件存在氢,以及ZN离子的名字不对,去掉氢和修改ZN离子名字后,能够正常进行了。
- #保存拓扑力场文件和坐标文件:目的为了mmgbsa计算做准备
- saveamberparm com native.prmtop native.inpcrd
- #将H、connect信息均删除
- #溶剂化,加盒子
- solvatebox com TIP3PBOX 10
- #检查电荷
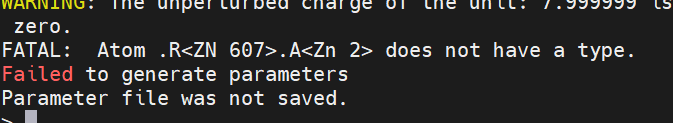
- charge com
charge com 是检查复合物体系带电情况,用于后面添加电荷,使体系为电中性,计算电荷结果:氯离子是64个,为了保证中和体系,添加70个Na+,64个Cl-
- (添加正负电荷的数目需要根据盒子的体积进行计算,使得最后的离子浓度为0.15M,同时也要保证体系为电中性)
- #加正负电荷
- addionsrand com Na+ 70 Cl- 64
- #再次检查电荷为0:
- charge com
- #保存蛋白质受体的拓扑文件和坐标文件
- saveamberparm pro anti.prmtop anti.inpcrd
- #保存配体的拓扑文件和坐标文件
- saveamberparm lig rbd.prmtop rbd.inpcrd
- #保存蛋白质和配体复合物的拓扑文件和坐标文件
- saveamberparm com complex.prmtop complex.inpcrd
- #保存蛋白质和配体复合物的pdb文件
- savepdb com com-min1.pdb
- ctrl+C退出tleap
- 一般使用tleap处理之后的结构,会自动对残基进行重新编号,这里导出pdb是为了检查重新编号的情况,后面的步骤会用到。
- 模拟小组公共空间上传了后面用于能量最小化等步骤的四个.in参数文件,链接如下:AMBER-MD参数文件
- #能量最小化
- pmemd.cuda -O -i min.in -p complex.prmtop -c complex.inpcrd -o min.out -r min.rst -ref complex.inpcrd
- min.in中的参数,重点关注:
- cut=10.0 (cut指cutoff,一般用10.0埃米,所有.in参数文件中尽量保持一致)
- restraintmask=':1-439' (这里的1-439修改成你的体系复合物实际的残基数目编号)
- #能量最小化
- pmemd.cuda -O -i min.in -p complex.prmtop -c anti.inpcrd -o min.out -r min.rst -ref anti.inpcrd
- min.in中的参数,重点关注:
- cut=8.0 (cut指cutoff,一般用10.0埃米,所有.in参数文件中尽量保持一致)
- restraintmask=':1-439' (这里的1-439修改成你的体系复合物实际的残基数目编号)
- 格式后查看:
- cpptraj -p anti.prmtop -y min.rst -x min.rst7
- ambpdb -p anti.prmtop < min.rst7 > min.pdb
- # 升温
- pmemd.cuda -O -i heat.in -p complex.prmtop -c min.rst -o heat.out -r heat.rst -ref min.rst
- 是内存被占满了,换了208,但还是报错,怀疑从能量最小化开始,可能就有问题,将min.rst文件转成PDB,发现还是体系有问题,蛋白质有断裂,体系崩溃了,与老师交流后,选择不加Zn离子,因为它里口袋很远,也不是发挥功能所必须的,而且之前彭师兄跑MD额时候就没有加Zn,那4个CYS都跑开了
- 注意检查和修改的部分同min.in
- # 预平衡
- pmemd.cuda -O -i density.in -p complex.prmtop -c heat.rst -o density.out -r density.rst -ref heat.rst
- 注意检查和修改的部分同min.in
- # 提交MD,注意调整md.in文件里的nstlim
- nohup pmemd.cuda -O -i md.in -p complex.prmtop -c density.rst -o md.out -x md.crd -r md.rst -ref density.rst &
- #续跑:
- nohup pmemd.cuda -O -i md.in -p complex.prmtop -c md.rst -o md_2.out -x md_2.crd -r md_2.rst -ref md.rst &
- restraintmask=':ZN,167,170,218,220' (这部分用于位置限制的残基修改为ZN2+以及与其形成配位键的残基)
- nstlim=100000000, (步长,该参数决定要模拟的时长,参考手册进行调整)
- cut=10.0, (与min.in, heat.in, density.in保持一致,建议统一用10.0)
- 后边采用了不加Zn离子的蛋白质,restraintmask=':ZN,167,170,218,220'和ntr那两行删掉才正常运行
对轨迹进行处理:去水、去周期性
- 用cpptraj处理:启动cpptraj
- cpptraj
- #读入拓扑及轨迹文件
- parm complex.prmtop
- trajin md.crd
- strip :WAT,Na+,Cl-
- autoimage
- 根据条件输出轨迹:只取1帧
- outtraj look.pdb onlyframes 1
- #导出能量分解所需要的轨迹文件:
- trajout md-nowat_lipid_200ns.nc
- run
- quit
- 用cpptraj处理:启动cpptraj
- cpptraj
- #读入拓扑及轨迹文件
- parm complex.prmtop
- trajin md.crd 0 5000 10
- strip :WAT,Na+,Cl-
- autoimage
- #导出能量分解所需要的轨迹文件:
- trajout md-nowat_lipid_200ns.pdb
- run
- quit
- cpptraj
- #读入拓扑及轨迹文件
- parm complex.prmtop
- trajin md.crd
- strip :WAT,Na+,Cl-
- autoimage
- #计算rmsd值:
- rms first !@H= out rmsd1.xvg
- #导出能量分解所需要的轨迹文件:
- trajout md-nowat.crd
- #trajout md-nowat_lipid_200ns.nc
- run
- quit
RMSD值计算:
先计算RMSD值,找出稳定的那一段进行能量分解
- #启动cpptraj
cpptraj - #读入拓扑及轨迹文件
- parm native.prmtop
- trajin md-nowat.crd
- #指定RMSD的计算对象:
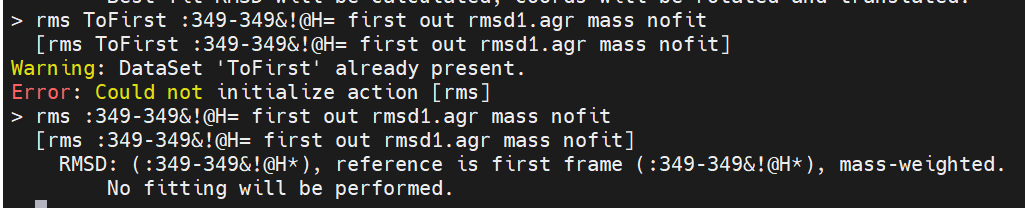
- #rms first :1-347&!@H= out rmsd1.xvg
- rms first !@H= out rmsd1.xvg
- #这里的1-347是指拓扑文件中的氨基酸残基编号
- #rms :348-348&!@H= out rmsd1.dat nofit
- #去掉Tofirst就可以了
- run
- quit
- 查看结果:直接用rmsd1.agr文档里的x,y坐标作图
MMGBSA能量分解
- 提交命令进行mmgbsa,但需要提前准备好mmgbsa.in文件:
- nohup mpirun -np 16 MMPBSA.py.MPI -O -i mmgbsa.in -o mmgbsa.dat -cp native.prmtop -rp anti.prmtop -lp rbd.prmtop -y md-nowat_200ns.nc -eo mmgbsa.csv &
- #mmgbsa.in的内容如下所示:
- Input file for running PB and GB
- &general
- startframe=1000, endframe=5000, interval=10,
- #问题1:怎么确定从哪一帧开始,到哪一帧结束,有判断依据吗?还有每隔几帧取一帧出来计算是要怎么判断啊
- #答:计算一下RMSD,取稳定的那一段计算,每隔几帧比较主观
- keep_files=0, debug_printlevel=2
- /
- &gb
- igb=5, saltcon=0.150
- #问题2:igb是用默认的5吗?,这里的摩尔浓度的盐浓度是用我MD时计算离子个数的0.15mol吗
- #答:igb一般用2或5,没有太大影响,摩尔浓度的盐浓度是0.15
- /
- &decomp # 计算分解自由能
- idecomp=1, print_res='1-121; 122-435', csv_format=0,
- #问题3:print_res=是不是改成我自己的‘蛋白质序列号’;‘配体序列号’
- idecomp和csv_format两个参数不变就可以了吧
- #答:print_res=这个参数可以删掉,这样系统会自动计算并列出所有的氨基酸残基的情况
- /