
rbd-ace2
ACE2体系准备
用/home/chpeng/zjfang/rbd-ace2/路径下准备好二硫键信息的ace2_H.pdb、rbd_H.pdb.
将ace2_H.pdb中的前19个氨基酸和第20位的H删掉。
- pdb2pqr30 rbd.pdb rbd.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output rbd_H.pdb
- source oldff/leaprc.ff14SB
- loadAmberParams frcmod.ions234lm_1264_tip3p
- loadAmberParams frcmod.ionsjc_tip3p
- pro=loadpdb ../ace2_H.pdb
- lig=loadpdb ../rbd_H.pdb
- bond pro.133.SG pro.141.SG
- bond pro.344.SG pro.361.SG
- bond pro.530.SG pro.542.SG
- bond lig.336.SG lig.361.SG
- bond lig.379.SG lig.432.SG
- bond lig.391.SG lig.525.SG
- bond lig.480.SG lig.488.SG
- com=combine {pro lig}
- saveamberparm com native.prmtop native.inpcrd
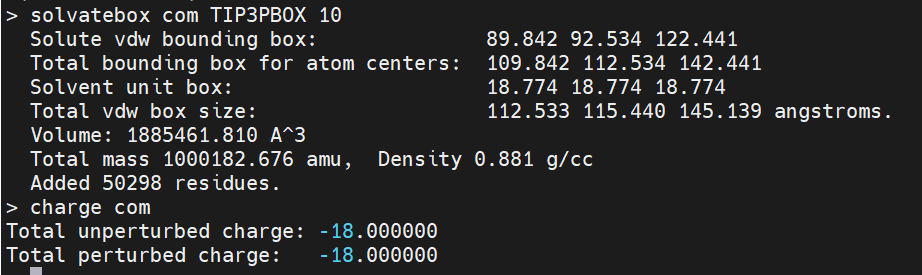
- solvatebox com TIP3PBOX 10
- charge com #计算0.15M的NaCl中和离子
- addionsrand com Na+ 189 Cl- 170
- #用这个公式算需要加多少Cl- https://ambermd.org/tutorials/basic/tutorial8/index.php
- #需要170个Cl-
- charge com
- saveamberparm pro ace2.prmtop ace2.inpcrd
- saveamberparm lig rbd.prmtop rbd.inpcrd
- saveamberparm com complex.prmtop complex.inpcrd
- savepdb com com-wet.pdb
- quit
转为GROMACS参数
- mkdir gmx
- cd gmx
- cp ../../../change.py ./
- python change.py
- #Amber参数转gromacs
- #Parmed (acype转过去之后,离子识别有点问题)
- #Python change.py
- import parmed as pmd
- amber=pmd.load_file("../complex.prmtop","../complex.inpcrd")
- amber.save("gmx.top")
- amber.save("gmx.gro")
- exit()
转为GMX参数后Zn2+被分成一个单独的moleculetype,但是限制的选项只能针对同一个moleculetype下的原子,所以需要把Zn2+修改到对应的moleculetype下。
好在离子不与其他原子成键,只需要在[ atoms ]的最后一行添加即可
可搜索[ bonds ],[ bonds ]的前面就是atoms的最后一行
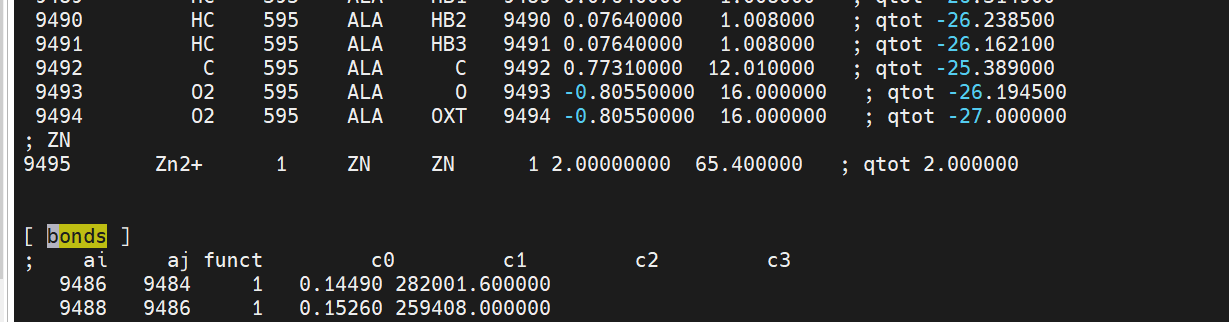
注意事项:既然已经把ZN合并到system1,在最后一项中的[ molecules ]中要记得删掉ZN 。除新添加的,所有ZN相关的条目都删除。
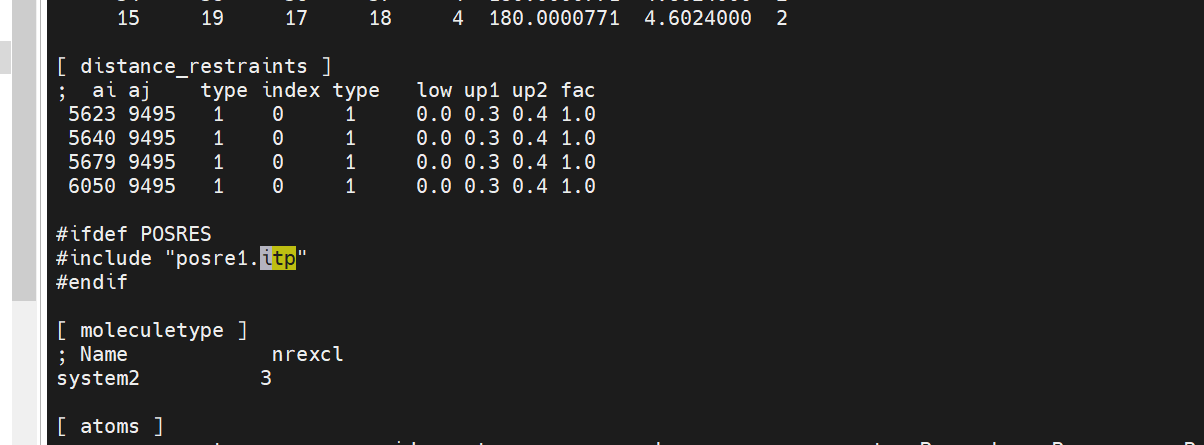
Zn2+的距离限制
gmx.top中添加如下信息:
- [ distance_restraints ]
- ; ai aj type index type low up1 up2 fac
- 5623 9495 1 0 1 0.0 0.3 0.4 1.0
- 5640 9495 1 0 1 0.0 0.3 0.4 1.0
- 5679 9495 1 0 1 0.0 0.3 0.4 1.0
- 6050 9495 1 0 1 0.0 0.3 0.4 1.0
- #ifdef POSRES
- #include "posre1.itp"
- #endif
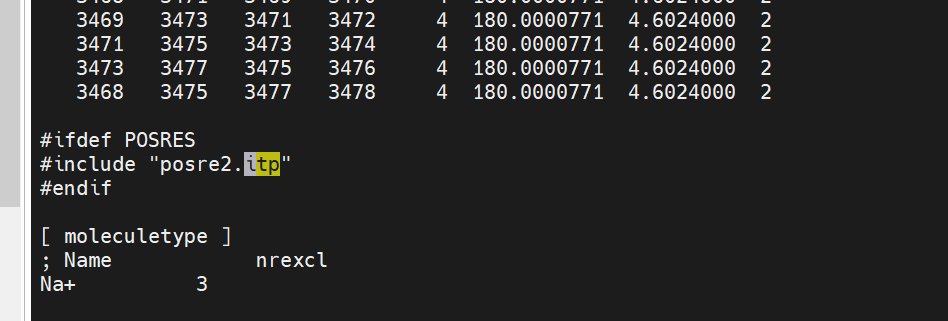
#这里的posre1.itp需要单独生成,对哪里限制就生成哪里的posre.itp文件。这里需要对两个蛋白质限制,所以生成两个,gmx.top文件中也需要添加两次itp
- #生成索引文件
- gmx_mpi make_ndx -f gmx.gro -o index.ndx
- #根据索引文件生成限制文件:选择protein-H,只限制重原子。
- gmx_mpi genrestr -f gmx.gro -n index.ndx -o posre.itp
- #也可以直接生成限制文件
- gmx_mpi genrestr -f gmx.gro -o posre.itp
注意事项:
1) posre.itp中的编号要与gmx.top中的一致,但是gmx.top中的原子编号与gmx.gro是不一致的
2)posre.itp需要预gmx.top放在同一个文件夹中
Gromacs能量最小化 :对于GMX2019,在进行grompp的时候一定要加 -r 选项
- #gpu:
- gmx_mpi grompp -f ../../em.mdp -p gmx.top -c gmx.gro -o em.tpr -maxwarn 1 -r gmx.gro
- nohup gmx_mpi mdrun -v -deffnm em &
- #CPU
- nohup mpirun -np 20 gmx_mpi mdrun -v -deffnm em &
Gromacs平衡: 需要一直平衡到dt=0.002不出问题
- gmx_mpi grompp -f ../../npt.mdp -p gmx.top -c em.gro -o npt.tpr -maxwarn 1 -r em.gro
- nohup gmx_mpi mdrun -v -deffnm npt &
npt不行,可以加 nvt调整密度:
- gmx_mpi grompp -f ../../nvt.mdp -p gmx.top -c npt.gro -o nvt.tpr -maxwarn 1 -r npt.gro
- nohup gmx_mpi mdrun -v -deffnm nvt &
npt、nvt来回几遍直到dt=0.002能跑
Gromacs提交md:
- gmx_mpi grompp -f ../../md.mdp -p gmx.top -c npt.gro -o md.tpr -maxwarn 1 -r npt.gro
- nohup gmx_mpi mdrun -v -deffnm md &
通过1ns的MD测试后,把md.tpr更改为200ns。超算上的版本是5.1.4 ,需要在cpu256上source到lywu.sh,再调用Gromacs延长时间到200ns
- source ~/lywu/lywu.sh
- mkdir tpr-200ns
- cd tpr-200ns
- gmx_mpi convert-tpr -s ../md.tpr -o md.tpr -until 200000
# convert-tpr:用于准备tpr文件,具体指延长MD模拟时间(通常该任务由于某些原因已经停止)时所需要的输入文件,即mdnew.tpr
#-s:指定已存在的tpr文件
#-f:指定轨迹文件
#.cpt:模拟断点文件(check point),该文件在模拟过程中每隔固定的时间间隔产生,保存了模拟系统的所有信息。若模拟因某些原因中断,可以使用该文件重新在断点处开始模拟,也可以从该断点文件开始,延长模拟时间
#-extend:指定延长的时间,单位ps,比如40000指40ns
只需要提供给超算平台md.tpr文件即可,任务运行命令如下
- gmx_mpi mdrun -v -deffnm md