PDBBind 使用的是中性条件 “For convenience, the “standard” protonation states at neutral pH were applied to the amino acid residues on the protein side. Thus, Asp, Glu, and His residues were negatively charge
GLN110,TYR268 突变实验 TYR268 突变实验(Y268A)有被报道 Schaftoside inhibits 3CLpro and PLpro of SARS-CoV-2 virus and regulates immune response and inflammation of host cells for the treatment of COVID-19 - Scienc
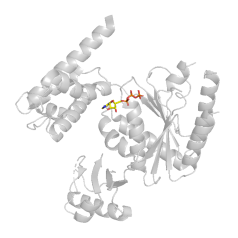
从PDB网站中查看该结构具体信息 发现该体系中存在一个非标准氨基酸CSX,无二硫键 判断非标准氨基酸是否需要处理 非标准氨基酸的一些考虑: 该氨基酸是否是结晶所需?若是,AMBER tutorials建议将其转换为标准氨基酸”you need to modify the resulting gfp.pdb file to convert the MSE residues (selenomet