
ONIOM优化(QM/MM)
1.蛋白前处理
根据韩子涧师兄的笔记ONIOM优化(QM/MM)和我做实验遇到的问题二创(2026.01.29),斜体是我增加的内容
PDB网站获得的结构往往发生残基缺失,需要进行补全,或者使用同源建模的结构。尽管同源建模和晶体结构RMSD很小,实际上有很多残基都会发生一定角度的偏离,有时可能会造成结果的定性错误。因此补全晶体结构的好处是结构更准确,但操作繁琐。使用同源建模操作简便,但可能带来误差。在实际情况中,有些问题可以使用同源建模(比如根本没解析晶体结构,就是希望引入偏差),有些问题则更建议使用晶体结构。
1.1补全侧链
1.1.1同源建模
这个和底下1.1.2补全晶体选一个做就好了(我用的是同源建模)
以PDB结构作为模板,生成残基完整的结构。
在PDB下载结构,FASTA序列。删去蛋白结构中所有配体(一定要去除配体),cofactor,水(这个可以用maestro的蛋白准备进行处理也可以PyMol手动删,如果有重要水可将其另存为新文件,后面会用),离子。多聚体可只保留一个单体。得到3RX4_pro0.pdb
如果有晶体结构,右侧选user template(以上传的结构为模板,对序列建模),如果没有晶体结构选sequences(检索数据库,进行匹配建模)。上传所需文件。
完成后下载,命名为3RX4_swissmodel.pdb。
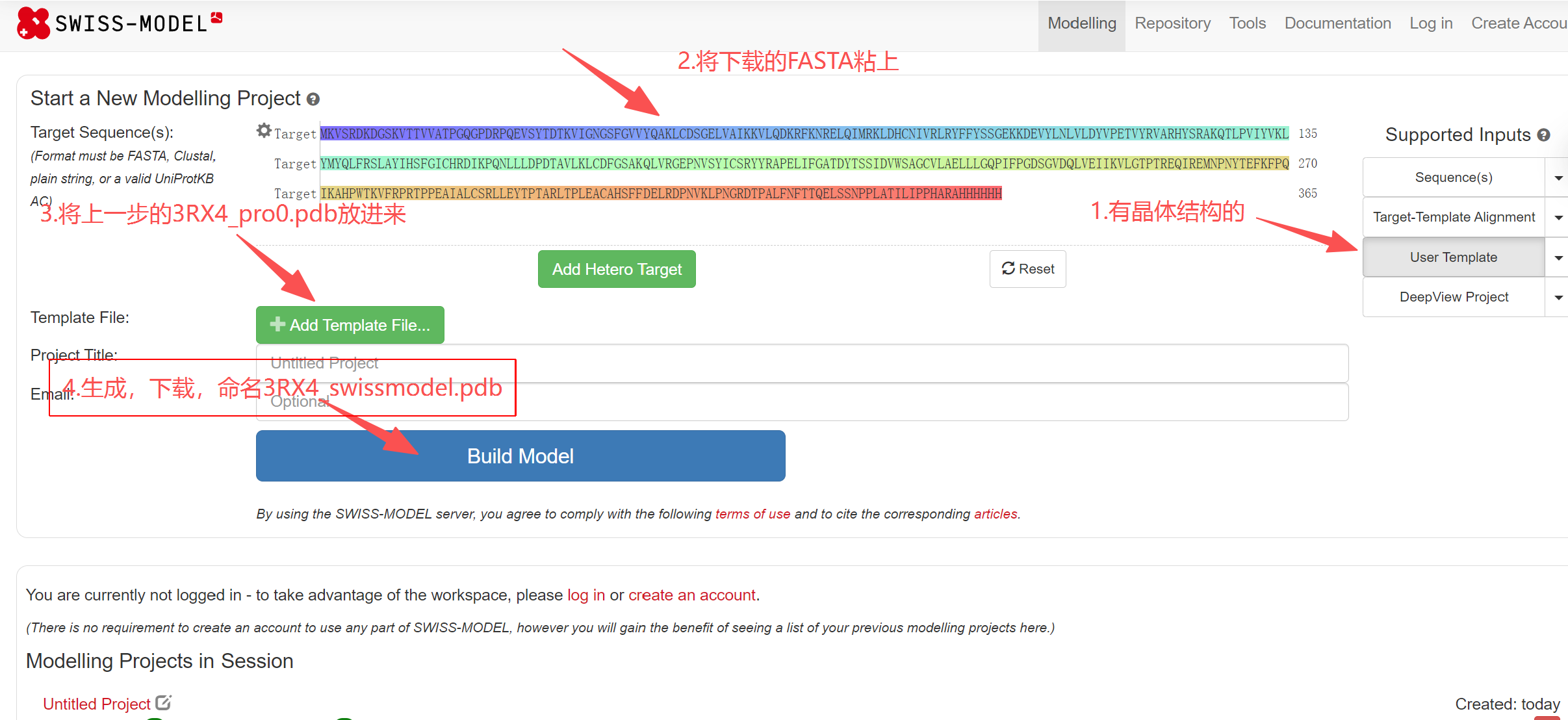
pymol加载建模结构及晶体结构,建模结构align to 晶体结构,保存为3RX4_swissmodel_align.pdb。建议观察下建模结构和原结构能否几乎完全重叠。(这一步是观察已有的结晶蛋白和建模出来的差别不太大)
1.1.2补全晶体
依据同源建模结构,手动补全晶体结构缺失的残基和原子。
文本打开晶体结构,检索missing,可以找到缺失残基和原子的说明,在swissmodel_align.pdb中找到对应的残基或原子,复制到晶体文件对应部分。一般情况下两端的残基可以不补,甚至可以删去一些。
1.2 蛋白质子化
1.2.1 pdb2pqr
这个也是和下面的选一个就行,我选的这个,我用的是172.21.85.11 g16 dddc1201
pdb2pqr(已知202有,不建议用新版的pdb2pqr30,因为老版的输出pqr文件中有蛋白电荷信息,后续需要用到)
- pdb2pqr --ff=amber --ffout=amber --chain --with-ph=5.0 3rx4_com.pdb 3RX4_pro_fix.pqr
设置的力场是amber,--chain为保留原始的原子编号(不从1开始,如果不设置,默认从1开始),--with-ph为结晶状态下的pH值(可以在pdb bank或者文献中查到),并自主处理末端氨基酸,即在C末端增加OXT(对于中间缺失的loop,不会增加OXT)。输出为*.pqr。
新版的pdb2pqr30调用(如cpu256上)
- conda activate pdb2pqr
- pdb2pqr30 input.pdb out.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output out.pdb
- 我用的是pdb2pqr_cli --ff=amber --ffout=amber --chain --with-ph=7.4 1wym_01.pdb 1wym.pqr
最后将pqr转化成pdb格式:
- obabel -ipqr *_pro_fix.pqr -o pdb -O 3RX4_pro_fix.pdb
1.2.2 H++网站
使用H++网站预测(没用过)
登录按提示操作即可
1.2.3 核酸质子化
pdb2pqr也可以处理RNA,但残基的名字需要改一下(For pdb2pqr, RNA resdiue names must be RA, RC, RG, and RU.):
在PyMOL中:
- alter polymer & resn A+C+G+U, resn = "R" + resn
obabel无法将RNA的pqr转换为pdb格式,无论名字是否是“R”+resn。可以将.pqr的后缀改为.pdb,然后读入PyMOL,存为pdb格式,再转化为obabel的pdb格式
- obabel -ipdb RNA_fix.pdb -opdb -O 3jbv_polymer_obabel.pdb
2.小分子前处理
动力学部分需要小分子的RESP拟合静电势电荷以及范德华作用,键角等信息,这些参数程序中并没有,因此需要自己生成。
2.1获得加氢PDB结构
原始文件 (一开始在结晶蛋白等的配体) 用pymol打开,将小分子保存为3RX4_lig.mol2。
01_keep3D.sh对小分子加氢
- chmod +x 01_keep3D.sh
- ../01_keep3D.sh -l *_lig.mol2
换pH需要手动修改01_keep3D.sh里面的-pH参数。如果结晶的pH和默认的相差不大可以不改。(也是pdb bank或者文献中的)
生成的一大堆文件只需要3rx4_lig_epikout.mae,pymol直接读取,必须再检查一遍结构是否正确,尤其是涉及胺的。(确定正确下面不用做)
不确定质子化是否正确的,可以再用maestro预测一次:加载3RX4_lig.mol2,右上角Tasks – Ligand Preparation – Generate possible states at target pH,输入结晶pH,+-后填0,最多生成2个结构即可,run。结束后在左侧选中生成的结构(要点圆点),观察质子化,不对的手动用GV补上。
pymol将小分子保存为3rx4_lig.pdb。
也可以使用下面的命令转,但是这样保存下来的pdb文件不包含小分子的名字。
- sdconvert -imae *lig_epikout.mae -osdf lig_epikout.sdf
- obabel -isdf lig_epikout.sdf -opdb -0 lig_epikout.pdb
GV打开pdb文件,保存为gjf文件3RX4_lig.gjf。
注意:若有重要的水分子,提取出来,加氢之后,保存,再和蛋白质及小分子共同存成一个pdb。最后用GW打开,生成优化文件。
2.2小分子参数计算
参考《量子化学-拟合RESP电荷、力场参数》 下面就是这个的内容,我粘贴了过来
1.Gaussian计算
需要准备的是小分子gjf文件(mol,sdf等,GV打开也可保存为gjf)
1.1传统gjf文件
下面是老版的,可以用,也可以用新版的,往后翻。
gjf文件加开头结尾,最终类似下面:
- %mem=4GB
- %nproc=4
- # opt hf/6-31g(d) geom=connectivity pop=mk iop(6/33=2,6/42=6,6/50=1)
- 3rx4_ligand
- 0 1
- C(PDBName=C4,ResName=SFI,ResNum=317_A) -8.34200000 9.07000000 17.71000000
- C(PDBName=C5,ResName=SFI,ResNum=317_A) -8.95900000 8.14900000 18.68000000
- C(PDBName=C6,ResName=SFI,ResNum=317_A) -5.62400000 6.49800000 18.05000000
- 1 15 1.0
- 2 23 1.0 24 1.0
- 3 5 1.0 7 2.0 10 1.0
- antechamber-ini.esp
- antechamber.esp
0 1:电荷、自旋多重度,按实际情况手动修改(可以关注O和N的价态),自旋多重度一般都为1。
opt:几何优化。非必需,但是一般都加。个人为加的原因是,RESP电荷充分考虑了分子柔性,因此在一定空间内与构象变化关系不大,优化可以将其他手段得到的结构(晶体/动力学等)进一步优化到构象空间内与之最相近的能量极小值点。
hf/6-31g(d):计算方法/级别。若需要溶剂,添加scrf=(solvent=xx)。考虑该参数时务必阅读4.。
geom:确保原子之间键连正确。加不加都行,理论上不影响结果,不加下面的数字键连都要删掉。
iop(6/33=2):进行RESP Fitting并输出到Gaussian的.log文件。
IOp(6/42):Density of points per unit area in esp fit. 0:Default (1) N:Points per unit area。
IOp(6/50):Whether to write Antechamber file during ESP charge fitting.
pop=mk:Produce charges fit to the electrostatic potential at points selected according to the Merz-Singh-Kollman scheme
1.2更新的gjf文件
我用的这个(Cl和Br)
- %mem=4GB
- %nproc=4
- # opt b3lyp/6-311g(d,p) pop=mk iop(6/33=2,6/42=6) em=gd3bj
- 3rx4_ligand
- 0 1
- C(PDBName=C4,ResName=SFI,ResNum=317_A) -8.34200000 9.07000000 17.71000000
- C(PDBName=C5,ResName=SFI,ResNum=317_A) -8.95900000 8.14900000 18.68000000
- C(PDBName=C6,ResName=SFI,ResNum=317_A) -5.62400000 6.49800000 18.05000000
- 只需要空两行以上就好,不用有尾巴了
其实原子后面的括号里的也可以去掉。不影响结果 (后面有遇到ResName=没有的情况报错,就直接去掉了)
em=gd3bj:色散校正。对于大部分没有分子内弱相互作用(氢键,卤键等)不加影响不大,但总的来说推荐加上。
注意:这里使用的是b3lyp/6-311g(d,p)计算级别是有争议的,传统的做法是使用HF/6-31G(d)真空。具体参考4.。
1ECV因为卤素是 I 的基组不同(参考罗睿童的,如下)
1ECV_ligand-log-2.gjf
- %mem=8GB
- %nprocshared=8
- #B3LYP/genecp em=gd3bj iop(6/33=2,6/42=6) opt pop=(MK,ReadRadii)
- 1ECV_lig
- -2 1
- C -0.89369700 -1.45633300 0.00074700
- C 0.49740400 -1.39470100 0.00086200
- C 1.17240100 -0.15687500 0.00041100
- ......
- C H O N 0
- 6-31g(d)
- ****
- I 0
- SDD
- ****
- I 0
- SDD
- I=2.3
- I=2.3
1.3提交Gaussian计算
将上述3RX4_lig.gjf加了相应基组和电荷信息后保存为3RX4_ligand.gjf
- nohup g16 3RX4_ligand.gjf &
如果是g09就写g09,最好用g16
2.手动提取参数
2.1提取小分子的电荷
我用的是手动,数据在85.23服务器上ID-antechamber文件夹里
- antechamber -i *.log -fi gout -o lig.prep -fo prepi -c resp
–nc -1
nc后面跟小分子的formal charge,但是貌似不用这个参数也可以。
antechamber生成的以ANTECHAMBER开头的文件中的原子顺序与gaussian输入中的gjf中的原子顺序一致,而生成的NEWPDB.PDB文件和.prep文件中的原子顺序与gjf文件中原子顺序不一致,因此将ANTECHAMBER_PREP.AC的电荷信息和原子类型输出即可。
- awk '$1=="ATOM" {print $3, toupper($10) "-" $9}' ANTECHAMBER_PREP.AC
得到:
原子名称 原子类型-电荷
N1 N3--0.297108
O1 O--0.703333
C1 C3--0.031802
复制粘贴保存为resp.txt文件,文件最后最好不要有空行,后续会用到。
2.2.提取力场信息
- parmchk2 -i lig.prep -f prepi -o forcefiled.frcmod -a y
生成的ligand.frcmod文件中有力场参数信息(可用于分子力学/动力学模拟)。
分别输入下面的命令:
- awk '/NONBON/, /NR/' *mod | awk '{print "VDW", $1, $2, $3}'
- awk '/^BOND/, /ANGLE/' *mod | awk '{print "HrmStr1", $1, $2, $3, $4}' | tr '-' ' '
- awk '/^ANGLE/, /DIHE/' *mod | awk '{print "HrmBnd1", $1, $2, $3, $4, $5}' | tr '-' ' '
复制粘贴保存以上命令产生的分子力场信息,其中可能会产生一些只有VDW或者其他没有数字的行,不需要。后续会用到。
3.自动提取参数
参考《量子化学-ONIOM自动化脚本》
4.计算级别和溶剂的考虑
传统的是在真空中使用HF/6-31G(d),是因为此级别会高估偶极矩10%-15%,正好等效反映出水的极化作用。此外,蛋白的分子力场参数拟合时,也是在这个级别下进行的,所以保持一致比较好(之前的文章基本都是这么做的)。但是,但这种高估效果并不稳定。近几年的文章使用b3lyp/6-311g(d,p)在隐式溶剂模型中计算的在不断增多。
卢天认为,传统级别在部分体系会造成较大的误差,可以使用更高级的方法进行精确计算(结合隐式溶剂模型)但是一旦结合溶剂,计算值将随着溶剂极性增大而越来越偏离实验值。比如,后续模拟是在水溶液中进行,这里也结合水模型计算,将导致RESP电荷异常,最终结果异常(因为水的极性太大了)。
综上,个人认为使用b3lyp/6-311g(d,p)。
a.如果用于ONIOM,建议使用真空。因为ONIOM就是在真空中进行的,而且对于大部分体系而言,量子化学优化几何结构时,真空中的构象和溶液中的相差不大。
b.如果用于已经考虑溶剂极化的力场,不需要使用溶剂,因为力场已经考虑这方面的信息,不需要通过原子电荷体现溶剂化效应。
5.参考
3.复合物处理与任务提交
将加氢的蛋白和配体在坐标上合并,同时根据研究所需,将原子换划分到高低层(QM和MM区),生成运行文件。
先合并再修改所用的基组信息+上述小分子的resp.txt文件+复制粘贴保存以上命令产生的分子力场信息(VDW 、HrmStr1和HrmBnd1)+修改空行 最后提交
3.1合并蛋白和配体
合并(若之前提取出水分子,同法添加):
- obabel -ipdb *_pro_fix.pdb *_lig.pdb -opdb -j -O 3rx4_com.pdb
3.2设置高低场
如有必要在pymol里先看好要选高层的原子
GV打开3rx4_com.pdb,tools-atom list,把需要的原子设置成high。
涉及残基的高层,在断键(选择高低层分界处)需要参考以下原则:
a.不可以切断双键(酰胺键具有双键性质,不可以断),环
b.断键处距离研究的原子至少两根键的距离
c.一般较少切断极性键(键的两端是不同原子就是极性原子),但不是不可以切,只是尽量
d.如果条件允许,可以把涉及残基的侧链全包括进高层,不然后面补参数麻烦。
tips:atom groups-onion layer可以查看高底层。
详细界面参考下图们:
图一:
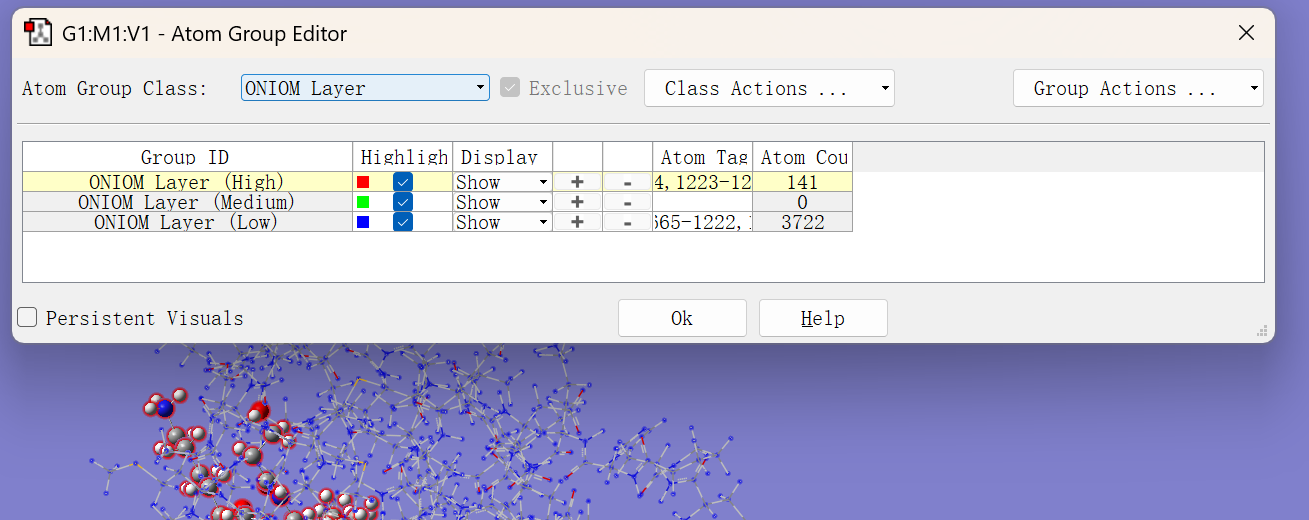
图二: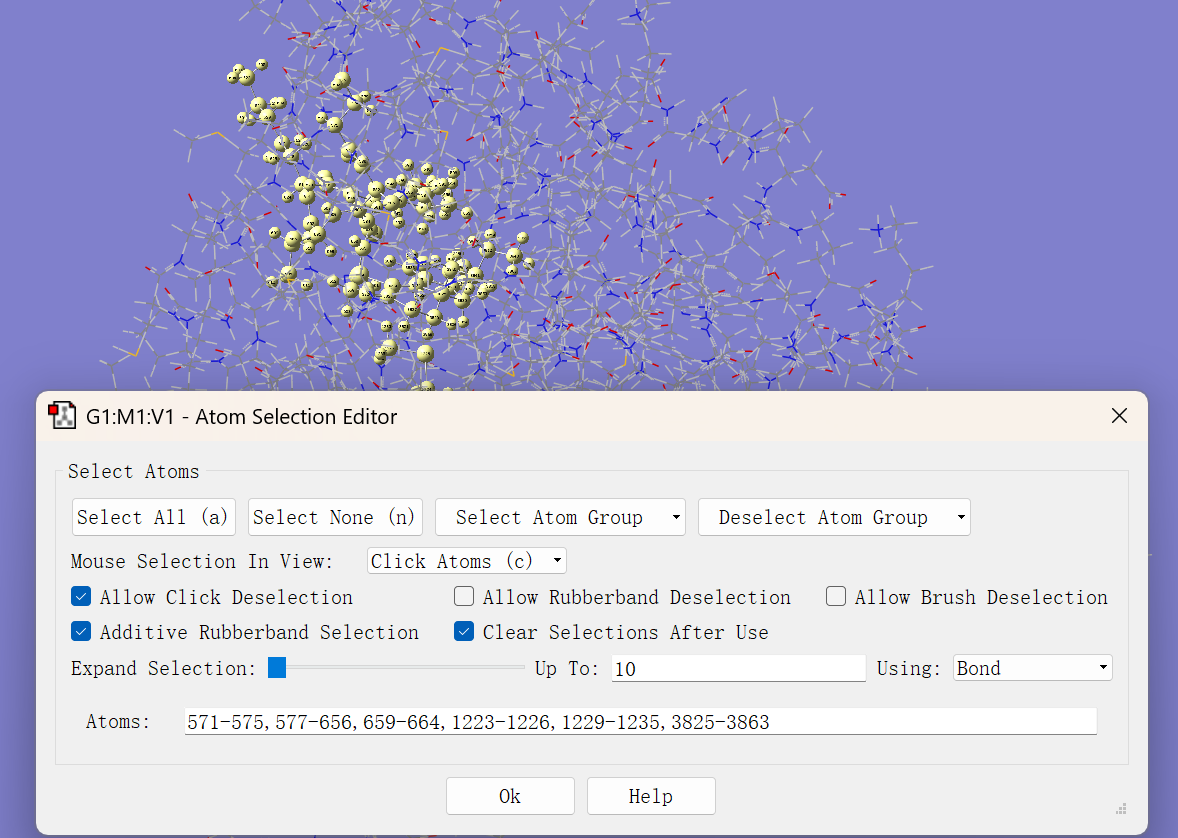
可以看一下底下Atoms是不是自己选的那些,如果不是可以在下图这个地方选中,然后在图一的高层点一下+
3.3生成gjf文件
Calculate - Gaussian Calculation Setup…
Job type: Optimization
Method: 右上角Multilayer ONIOM Model;
High layer: DFT - B3LYP -6-31G(d), charge根据具体体系定,保证spin(多重度)为1
Low layer: Mechanics - Amber,查看pro_fix.pqr文件中的电荷,如果高层包括了带电残基,对应低层需要做相应扣除,保证spin(多重度)为1。注意,这里设置的是整个体系的电荷。
Title:自定
Link: Chk file(specify, 前面的路径都去掉,取消勾选full path); Memory Limit,Shared Processors自定 (推荐核数:内存GB=1:1,16核)
General:可以把geom取消勾选,但是geom=connectivity理论上是不影响结果的。
下面点Retain,save gjf(要勾选Write Cartesians,)。
注:按照正确的方式选择电荷后,多重度为1,检查:
a.误选了一些孤立的原子到高层
b.配体是共价的(程序加氢的时候不会考虑是否共价,但是加上氢以后氢原子会和原来的残基上的原子发生部分重叠,导致自选多重度错误)
c.因其他原因造成的原子重叠
d.如果条件允许,可以把涉及残基的侧链全包括进高层,不然后面补参数麻烦。
根据d,其实能断键的地方就三个(下图供参考)
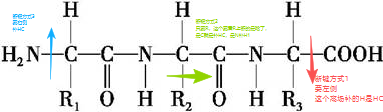
这个里面的电荷设置很重要:High layer的电荷是根据你选择的原子(残基,重点关注酸碱氨基酸和配体小分子的电荷),Low layer的电荷是整个体系的总的电荷
3.4补充电荷和力场信息
(1)小分子信息补充
找到复合物gjf文件中缺失电荷的地方,一般是在最后的配体,类似下面:(F及下面就没有了)

上图‘-’后面的原子类型不对,后面也没有电荷信息,需要依据2.3中的内容,更改这里的内容。理论上这里配体每个原子的顺序和resp信息中的原子顺序是一样的,可以使用下面的脚本快速生成:
- python lig_solve.py -g 3RX4/3rx4_oniom.gjf -r 3RX4/resp.txt
将输出的结果覆盖原gjf的相应部分即可。
这一部分与下面geom产生的键连信息之间有且只能有1个空行。(这个不知道为啥一行)
(20230921曹瑞妮报告脚本一处bug,已修复)
- H-HC(PDBName=H202,ResName=FBR,ResNum=1) 0 23.94300000 13.30400000 118.19900000 H
- H-HC(PDBName=H203,ResName=FBR,ResNum=1) 0 23.26500000 11.97800000 119.20400000 H
- 这个要两行空格
- VDW c3 1.9080 0.1094
- VDW hc 1.4870 0.0157
- 最后要2行或以上的空行
(2)高低层切换处原子信息补充
gjf文件中检索‘L H’(不包括引号)找到下面这样的地方:(这个-HC是我根据我自己的补的)

Gaussian会将高低层切换处的原子用H进行封闭,需要按照Amber Atom Types in General AMBER Force Field (GAFF)中的描述,找到对应原子类型,在H后补充该H的原子类型。
表格里提到的H在哪个原子上,看的是高低层切换处的高层原子。
注意:这里查看原子类型时,需要判断C上有几个吸电子基团,判断的标准不是常规的有机化学判断标准,而是看原子。比如补充C1上的H的原子类型,只要C1上没有除H,C以外的原子,这个C上就没有吸电子基团(哪怕C1上连着羰基)。(20230921曹瑞妮修正)
一般就两类:H1和HC,具体可以去上面网址看:

这个吸电子基一般指N原子

结合上面三种常见高层断键方式一起看
最后补完类似下面的:
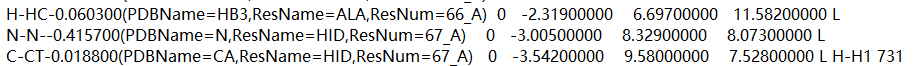
(3)在gjf文件最后打几个空行。(两个及以上)
(4)修改gjf文件开头
- %mem=16GB
- %nprocshared=16
- %chk=3rx4_oniom.chk
- # opt oniom(b3lyp/6-31g(d) em=gd3bj:amber=hardfirst) geom=connectivity
amber=hardfirst表示有程序预定义的参数就优先使用,没有再使用我们之前补充的参数。
由于gjf文件可能会缺少计算所需的参数,但是正常运行数十分钟才会报错,可以使用下面的关键词,先检查文件是否有问题,以节约时间
- %mem=16GB
- %nprocshared=16
- %chk=3rx4_oniom.chk
- # opt oniom(b3lyp/6-31g(d) em=gd3bj:amber=hardfirst)=onlyinputfiles geom=connectivity
=onlyinputfiles表示只读取信息并输出,不进行计算。
如果参数有问题,会报missing ...处理办法见3.5
如果参数没有问题,会报错:
FileIO operation on non-existent file.
去掉=onlyinputfiles,运行。
- nohup g16 3rx4_oniom.gjf &
3.5.缺失参数报错处理
missing atom parameter:
AMBER力场中缺失参数,可以现在Gaff力场文件中找一找,二者完全兼容
找到对应原子,看需要补充什么信息,复制到小分子的力场参数后面就可以。参数在202:$AMBERHOME/dat/leap/parm下
下面是最常用到的,甚至可以在3.4直接把这个复制过去进行文件检查:
HrmBnd1 hc c3 h1 35.0 109.5
HrmBnd1 hc ct h1 35.0 109.5
gaff.dat (vDW、键长、键角、二面角等)小分子的力场参数
parm99.dat(vDW、键长、键角、二面角等)蛋白的力场参数
FFRead: read line ""
Blank file name read.
具体原因未知,从PDB重新制作了一下gjf文件就可以了
也有可能没错但是中断了,即使nohup...&交到终端了,也要exit,安全退出。如果没报错但是中断了,重新交吧
4.后续检测和判断
一般来说log文件可能几个小时补更新,但是chk文件更新还是很快的,可以看一下
高层200个原子以内,在不报错的基础上,跑2周都算正常
最后结束可以tail查看有没有计算时间和Normal
- Job cpu time: 88 days 9 hours 38 minutes 20.0 seconds. Elapsed time: 5 days 17 hours 37 minutes 36.8 seconds. File lengths (MBytes): RWF= 2103 Int= 0 D2E= 0 Chk= 617 Scr= 1
- Normal termination of Gaussian 16 at Tue Jan 27 17:24:35 2026.
一般是先看跑完高斯后的log文件(用高斯软件打开),看一下跑完后选中的残基和小分子之间的键长键角的变化(因为我是分析卤键),然后可以存出pymol可以打开的文件类型再看看