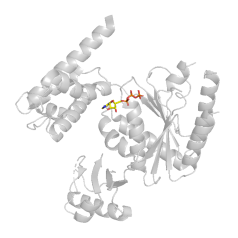
GLN110,TYR268 突变实验 TYR268 突变实验(Y268A)有被报道 Schaftoside inhibits 3CLpro and PLpro of SARS-CoV-2 virus and regulates immune response and inflammation of host cells for the treatment of COVID-19 - Scienc
从PDB网站中查看该结构具体信息 发现该体系中存在一个非标准氨基酸CSX,无二硫键 判断非标准氨基酸是否需要处理 非标准氨基酸的一些考虑: 该氨基酸是否是结晶所需?若是,AMBER tutorials建议将其转换为标准氨基酸”you need to modify the resulting gfp.pdb file to convert the MSE residues (selenomet