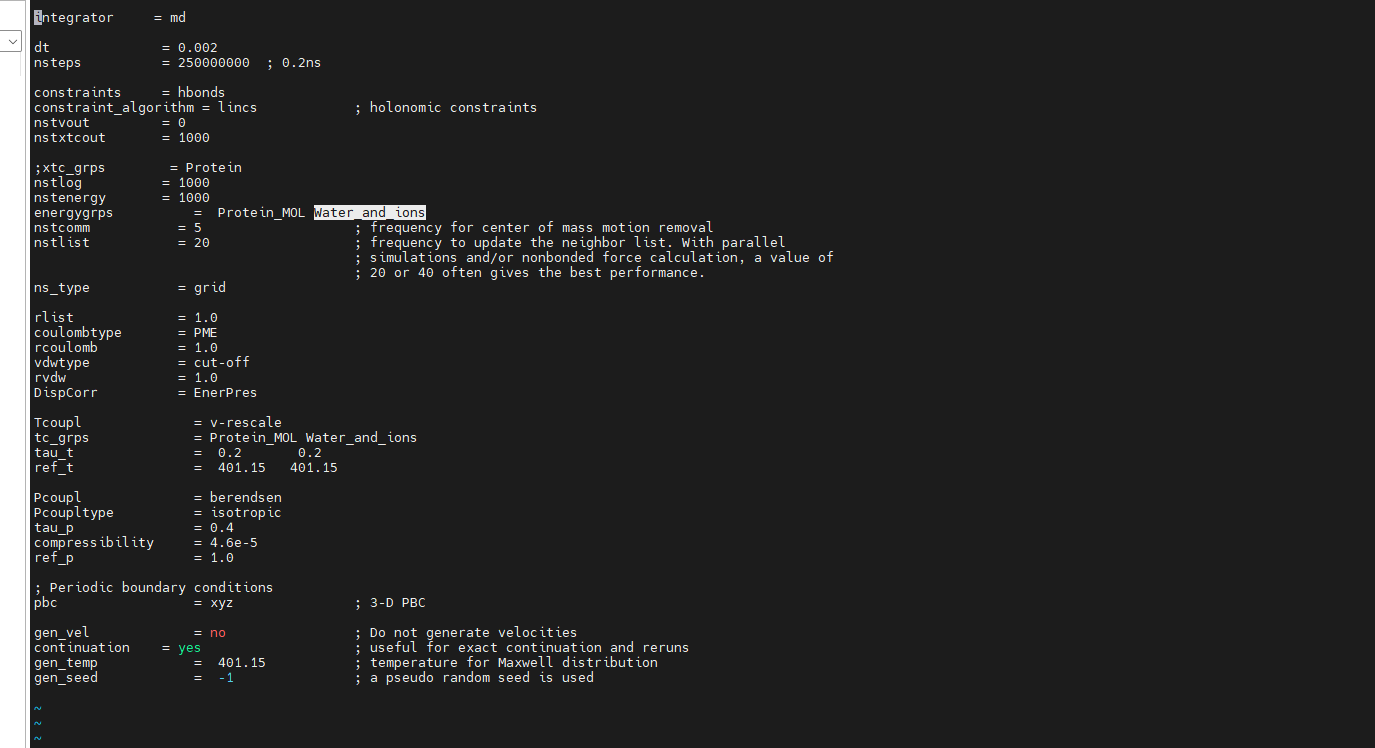
MD
配体准备
- # 22224
- # 172.21.85.24
- # 使用薛定谔优化结构(图形化界面操作)
- # pH = 8.0,手性从3D结构定(晶体结构),力场为2017版的默认力场OPLS3
- conda activate ambertools
- # 高斯优化
- # 注意检查体系的电荷和自选多重度(eg.将T3的gjf电荷0 1改为1 1)
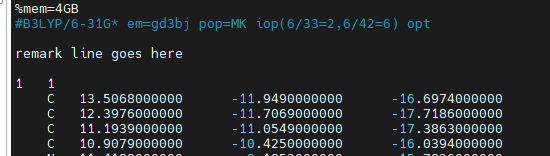
- antechamber -i lig.sdf -fi sdf -o lig.gjf -fo gcrt -at gaff2 -gn "%nproc=4" -gm "%mem=4GB" -gk "#B3LYP/6-31G* em=gd3bj pop=MK iop(6/33=2,6/42=6) opt" -rn MOL -nc 1
- -nc后面加电荷数,电荷为1就是1,电荷为-1就是-1,在gif文件里有体现
- 自旋多重度计算:(未配对的正电子数-未配对的负电子数)*2+1=1 一般都为1,遇到金属离子需要特别注意
- gif文件如下
- 第一个1指的是电荷数,电荷为-1就是-1
- 第二个1指的是自旋多重度
- #把gjf文件转成log文件,转换之前检查电荷和自旋多重度
- g16 lig.gjf
- nohup g16 lig.gjf &
- # 基于高斯优化日志文件,拟合RESP电荷,生成AMBER需要的参数文件(prep)
- antechamber -i lig.log -fi gout -o lig.prep -fo prepi -c resp;parmchk2 -i lig.prep -f prepi -o lig.frcmod -a y
- #这里会生成很多文件,包括NEWPDB.PDB
- 下面将进行两次对齐!
- 这一步是为了后面将坐标信息对其。注意:必须以高斯产生的小分子sybmol为基准,因为之前产生的键连信息lig.prep是高斯坐标为基准产生的。但我们需要的是对接的原子坐标信息,所以两次对齐的基准不一。第一次对齐(symbol):高斯产生的小分子为基准(NEWPDB.PDB)。第二次对齐(coordinate):对接的晶体结构为基准(crtstal.pdb)为基准。
- #删除H,获取坐标信息sdf
- gv里分别打开 NEWPDB.PDB 与 crystal.sdf(晶体结构配体文件):打开GV tools-atom list--鼠标点击symbol列-rows-sort selected -拖动鼠标选择所有H-edit-delete-selected atoms),接着仍在atom list中,edit-z matrix-standardize,分别保存。得到 gv-NEWPDB.pdb gv-crystal.pdb
- #第一次对齐
- GV里 检查 gv-NEWPDB.pdb gv-crystal.pdb,更改gv-crystal.pdb (atom list - Tag),使其Tag与gv-NEWPDB.pdb 完全一致。
- #第二次对齐
- dos2unix *
- grep -v "CONECT" gv-NEWPDB.pdb |cut -b 1-26 > symbol
- cat gv-crystal.pdb | cut -b 29-78|sed '1,2d'|sed '1i Combineing the coordinates from the crystal structure with the atomic numbers from the Gaussian optimized structure'> coordinate
- paste -d " " symbol coordinate|sed '1i TITLE lig.pdb'> lig.pdb
- ### 可视化检查
受体准备
- conda activate pdb2pqr
- pdb2pqr30 5xvg.pdb 5xvg.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output 5xvg_pred.pdb
- # 检查蛋白是否有二硫键 (pymol-show-disulfide, select resn CYS 检查位置)
- # 如果蛋白中存在二硫键,需要进行特殊的处理(教程待完善)
- pdb2pqr 是由APBS 项目开发的工具,APBS(Adaptive Poisson-Boltzmann Solver)用于分子静电势计算。主要用于添加氢原子、计算质子化状态,并根据残基的电荷和原子半径信息生成PQR 文件。PQR 文件是一个扩展的 PDB 文件,其中包含电荷和原子半径信息,这对于溶剂化自由能、静电势求解非常重要。
- pdb2pqr 的主要功能
- 添加缺失的氢原子。
- 计算特定 pH 条件下的质子化状态(如 ASP、GLU、HIS 等)。
- 生成 PQR 文件(带有电荷和原子半径的分子文件),适用于静电势求解器(如 APBS)。
复合物准备
- conda activate ambertools
- tleap
- source leaprc.protein.ff19SB # 加载蛋白质力场
- source leaprc.phosaa10 # 加载磷酸化氨基酸支持
- source leaprc.gaff2 # 加载配体力场
- source leaprc.water.opc # 加载溶剂力场
- # 导入配体参数
- loadamberparams"q/home/lywu/pengziyu/T3/lig_T3.frcmod"
- loadamberprep "/home/lywu/pengziyu/T3/lig_T3.prep"
- # 加载蛋白质和配体文件
- # 加载配体文件
- mol = loadpdb "/home/lywu/pengziyu/0103/ligand/lig-t28.pdb"
- # 加载蛋白文件
- pro = loadpdb "/home/lywu/pengziyu/0103/pre_protein/5xvg_pred.pdb"
- # 组装蛋白配体
- com= combine {pro mol}
- # 保存amber参数(不加溶剂的)
- saveamberparm pro pro.prmtop pro.inpcrd
- saveamberparm mol lig.prmtop lig.inpcrd
- saveamberparm com native.prmtop native.inpcrd
- #还没加水的prmtop
- savepdb com com-dry.pdb
- charge com #检查电荷
- solvatebox com OPCBOX 12.0 # 溶剂化和离子添加
- addionsrand com Cl- 0 # 添加离子中和体系
- # 保存拓扑和坐标文件
- saveAmberParm com complex.prmtop complex.inpcrd
- savePdb com complex.pdb
- quit
- # AMBER 参数转 GMX 参数
- #进入python
- import parmed as pmd
- amber = pmd.load_file('complex.prmtop','complex.inpcrd')
- amber.save('gmx.top')
- amber.save('gmx.gro')
- #给蛋白(所有的链)和小分子所有的链加上位置限制,都在[moleculetype]后加上(对gmx.top)进行操作
- ; Include Position restraint file(分号不能省略)
- #ifdef POSRES
- #include "posre1.itp
- #endif
生成限制文件
- source ~/.gmx2022.5-remd.sh
- # 生成位置限制文件,规定哪些原子被限制
- #限制蛋白
- gmx_mpi make_ndx -f gmx.gro(选择组生产index)
- gmx_mpi genrestr -f gmx.gro -o posre1.itp(分别选择蛋白)
- #这里选择group只需要选择被限制的那一组,譬如限制蛋白就只选择Protein,用于限制蛋白。
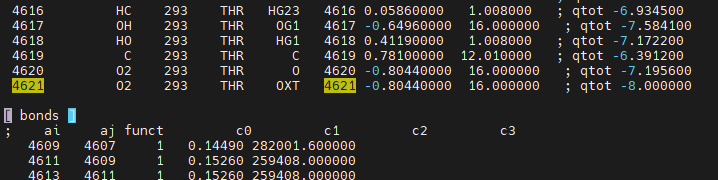
- #如果是多条链,需要查看第一条链的原子数(例如查询'[',找到[bond]上的第一列数字,如果是4621则删除posre1.itp后的所有的内容,并保存)
- #如果蛋白的两条链都是一样的原子数,那么两条链所用的位置限制文件可以是一样的,如果两条蛋白链原子数不一样那就要视情况而定。
- # 限制配体 (下面的57 替换为你的配体原子数+4,+4的目的是posre.itp前四行有固定内容要保留)
- sed -n '1,57p' posre1.itp > posre2.itp
体系平衡
- mkdir pre-equ
- 在pre-equ目录下mkdir em nvt npt mdp
- 一般预平衡跑200ps(dt=0.002;nsteps=100000)
- # 85.24
- cp /home/databank/lywu/vsremd_plus/protein-ligand/trypsin-benzamidine/pre-equilibrium/mdp-file/* ./mdp/
- cp gmx.gro gmx.top pre-equ
- ### 能量最小化 在em/
- gmx_mpi grompp -f ../mdp/em.mdp -c ../gmx.gro -r ../gmx.gro -p ../gmx.top -o em.tpr
- nohup gmx_mpi mdrun -v -deffnm em &
- ### 等温等容预平衡 在nvt/
- gmx_mpi grompp -f ../mdp/nvt.mdp -c ../em/em.gro -r ../em/em.gro -p ../gmx.top -o nvt.tpr
- nohup gmx_mpi mdrun -v -deffnm nvt &
- cd ../
- ### 等温等压预平衡 在npt/
- gmx_mpi grompp -f ../mdp/npt.mdp -c ../nvt/nvt.gro -r ../nvt/nvt.gro -p ../gmx.top -o npt.tpr
- nohup gmx_mpi mdrun -v -deffnm npt &
- # 检查 npt.gro 的构象
Production MD
- #生成index文件,选择group,退出q
- gmx_mpi make_ndx -f gmx.gro -o index.ndx
- #index文件是接着group生成的,可随意两两分组,但在GPU中无法识别,所以一般选择System;在CPU中可以识别可以自由选择组类别
- mkdir mdtest
- cd mdtest#准备一个用于分子动力学模拟的 tpr 文件。它结合了模拟参数文件、结构文件、检查点文件、拓扑文件和索引文件等信息,并生成最终的 md.tpr 文件。这个文件将会被用作后续 mdrun 模拟的输入。
- GPU 75.1
- source ~/.gmx-2022.5-GPU.sh
- export CUDA_VISIBLE_DEVICES=0
- gmx_mpi grompp -f ../mdp/md-1ns.mdp -c ../npt/npt.gro -t ../npt/npt.cpt -p ../gmx.top -n ../index.ndx -o md.tpr
- nohup gmx_mpi mdrun -v -deffnm md &
- CPU 85.24
- source ~/.gmx2022.5-remd.sh
- gmx_mpi grompp -f ../mdp-1/md-10ns.mdp -c ../npt/npt2.gro -t ../npt/npt2.cpt -p ../gmx.top -n ../index.ndx -o md.tpr
- nohup mpirun -np 100 gmx_mpi mdrun -v -deffnm md &
- gmx_mpi mdrun -s topol.tpr -deffnm output -nb gpu
可以暂时不做
- source ~/.gmx2022.5-remd.sh
- # 生成index (GMX 里没有 protein_MOL 这种现成的 group,我们可以通过index文件生成)
- # 例如我们想生成Protein_MOL 和 Water_Cl- 这两个组,可如下操作
- gmx_mpi make_ndx -f gmx.gro -o index.ndx
- > 1|15
- > 17|16
- # 注意,group序号以你自己的为准,上面的只是示例