
Glide虚拟筛选
1.概览
首先需要准备蛋白和配体/化合物库,包括补链,加氢,优化等。 如有晶体结构,首先进行对接复现晶体结构,以确定对接参数,再进行虚筛; 如果没有晶体结构,但是知道口袋位点,可以考虑直接虚筛,然后使用进行可靠性评价。不清楚口袋位点或是蛋白柔性比较大的情况,需要结合MD等工具先探明结构,过程复杂,以后可以单独开一章,所以这部分内容就不在本篇教程中包含了。
SPECS库的分子比较大,结构不好看,同样数量的分子对接较慢。 ChemDiv库分子数量多,但是结构整齐,对接起来较快。
虚筛时为了加快速度,可以先使用HTVS高通量,对打分较好的一批,如10万个分子或优于-7的,再次使用SP进行对接,后续甚至可以接着XP。
虚筛是为了寻找活性分子进行实验,得到先导化合物的。一般一次送样最多两百个化合物。
数据库拆分成了多个ligprep.maegz包,可根据大小给不同的CPU数量。
2.体系准备
2.1蛋白准备
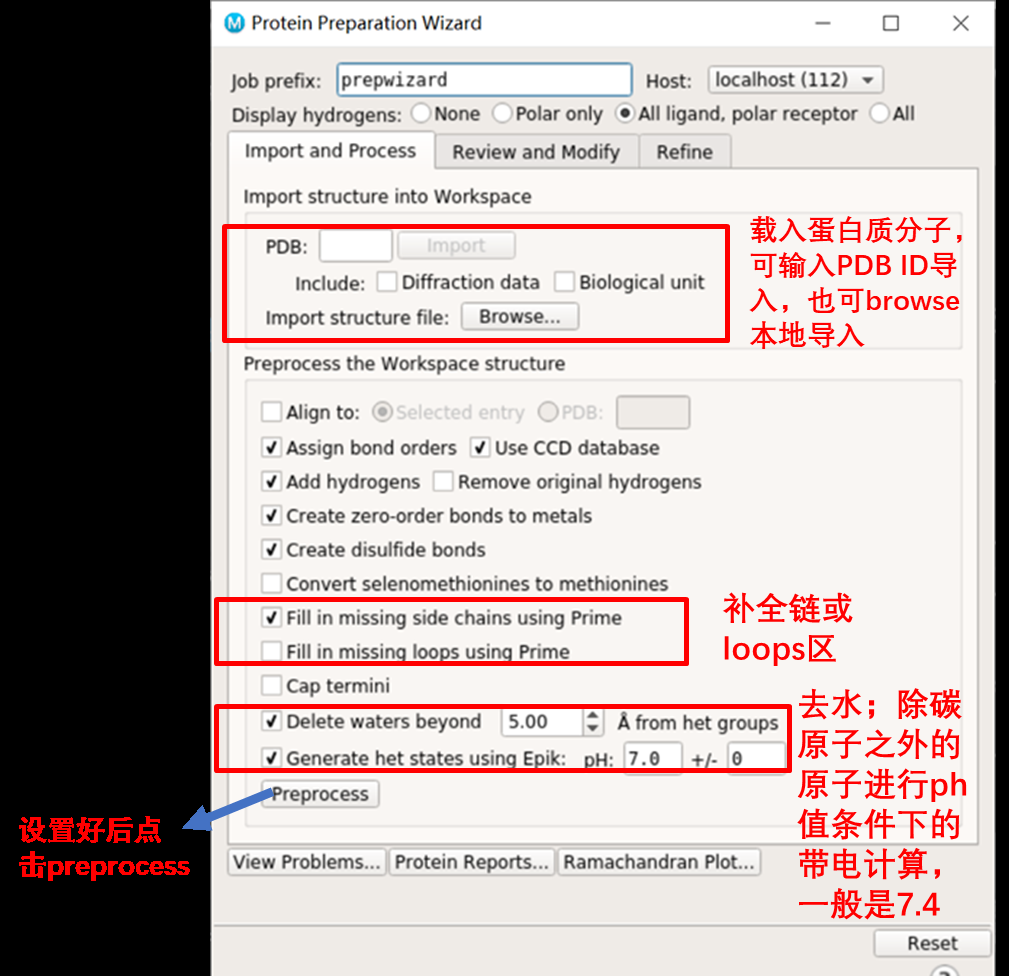
若存在问题,则会弹出Protein Preparation-problem窗口,包括四个问题选项:原子类型(Atom Types)、侧链丢失信息(Missing Atoms)、原子位置冲突信息(Overlapping Atoms)、原子坐标改变(Alternate Positions)。 若有侧链丢失信息问题可点击Add Missing Side Chains来添加丢失残基,然后点击OK即可。 若是发生问题的部分不在活性口袋周围,不改变也可。 另外,我们也可以点击View Problem来随时查看结构问题。
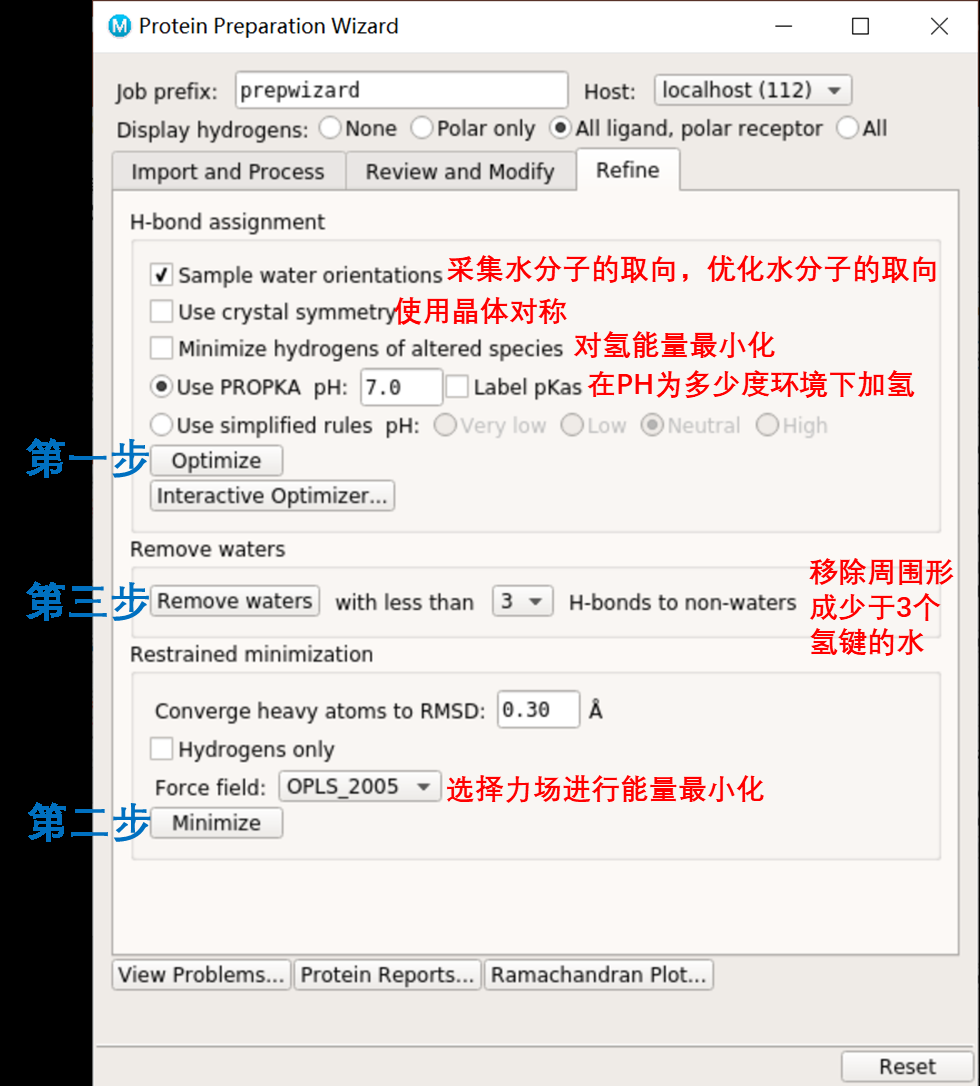
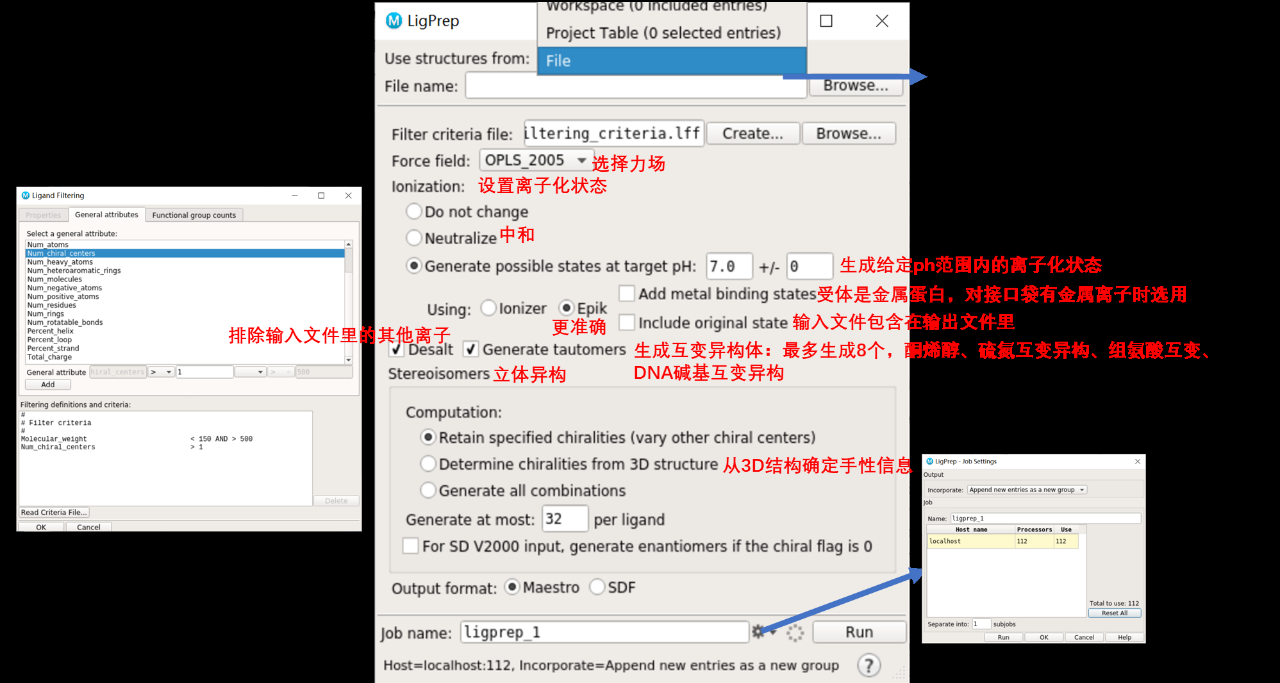
2.2配体准备
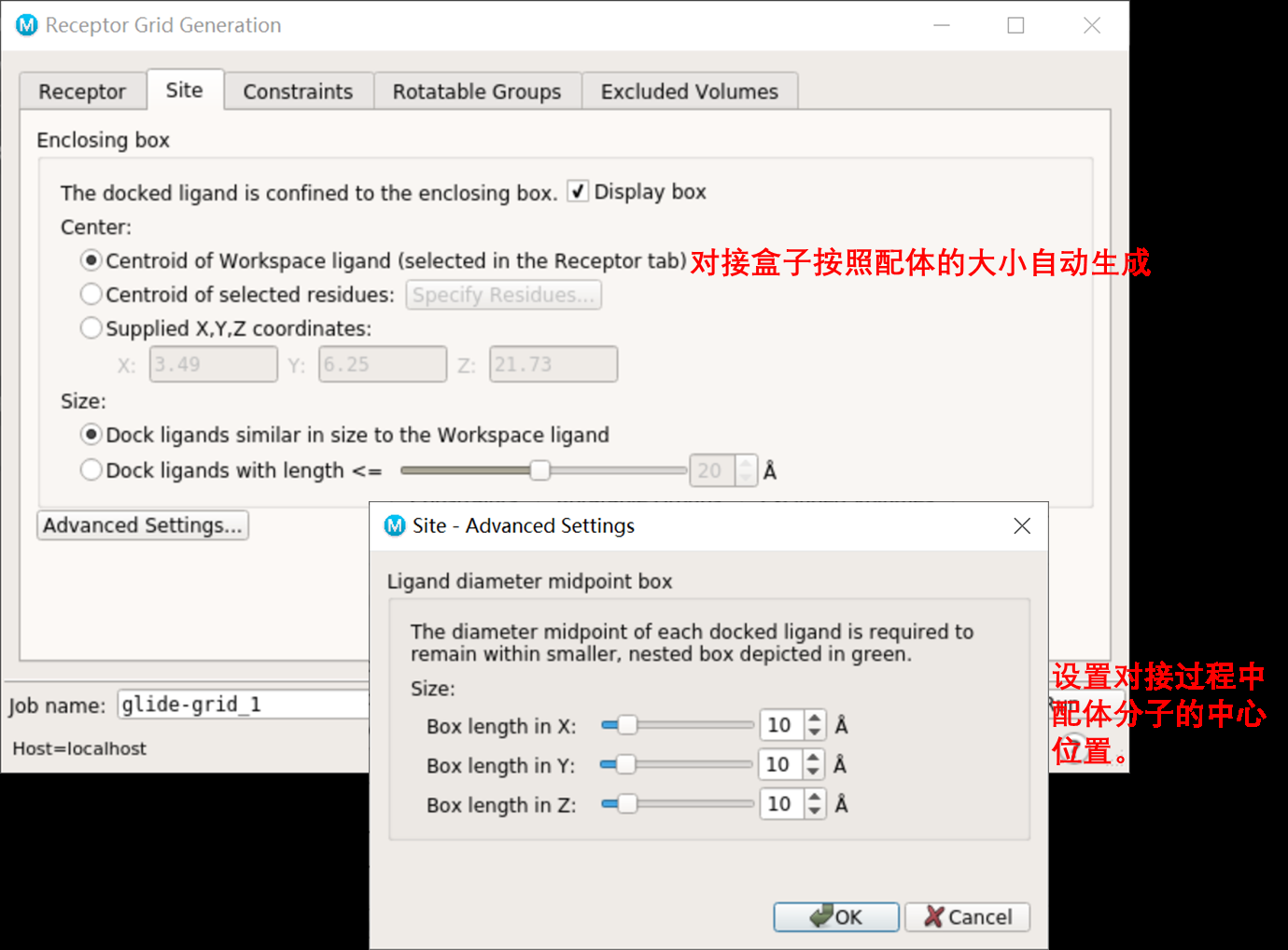
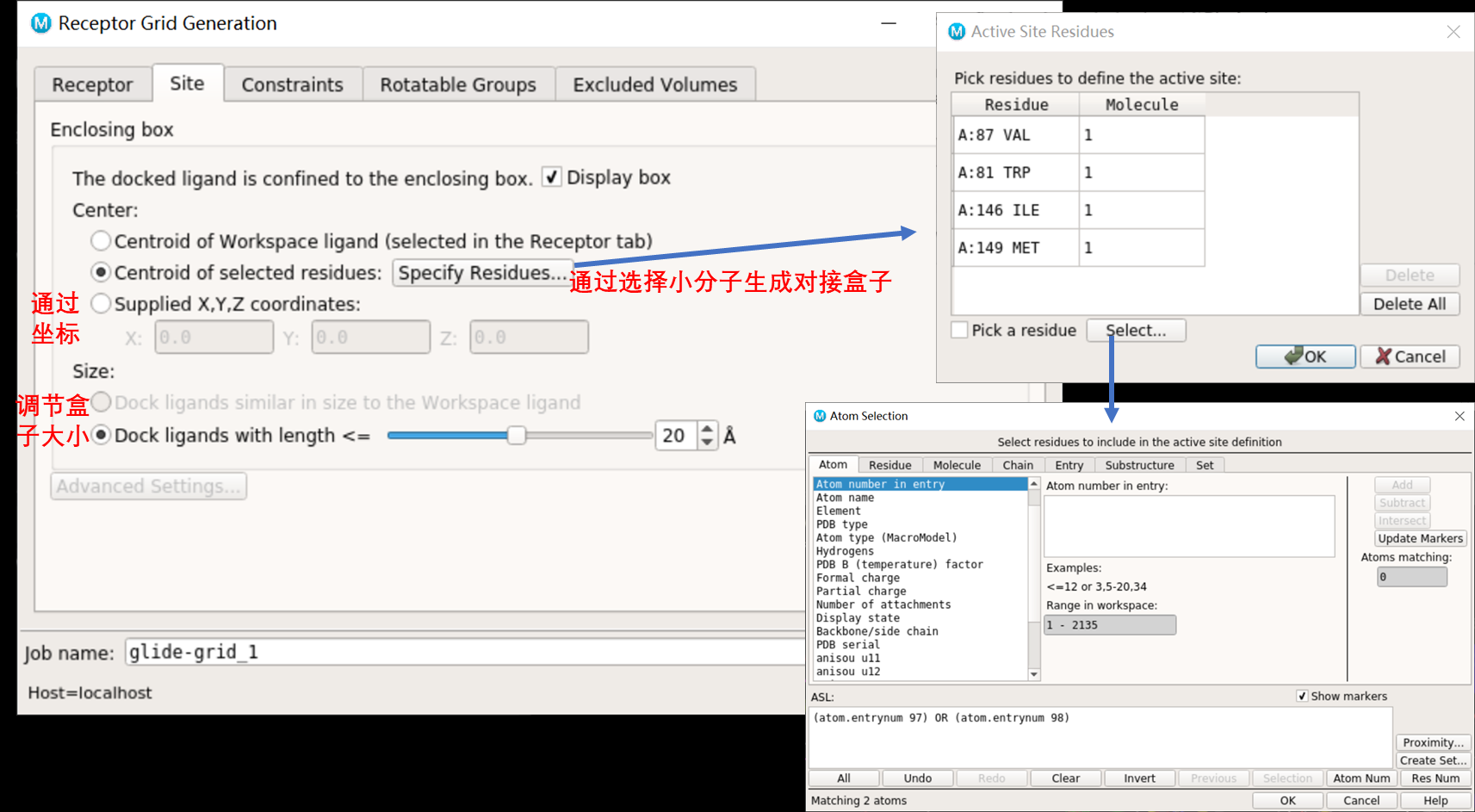
2.3生成对接盒子
一般只需设置receptor和site,用默认就可以。
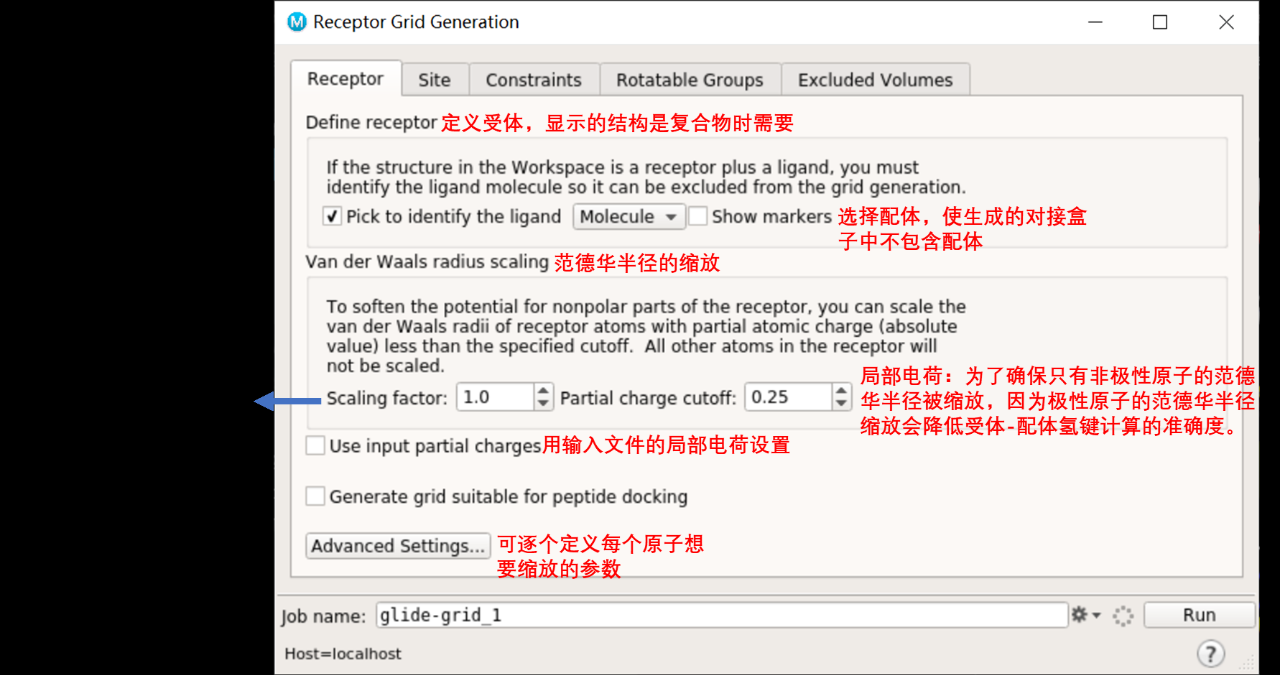
2.3.1有配体存在
不关闭这个界面,回到薛定谔主界面,左键选择配体,即可自动生成盒子。
2.3.2没有配体
2.3.4设置约束条件
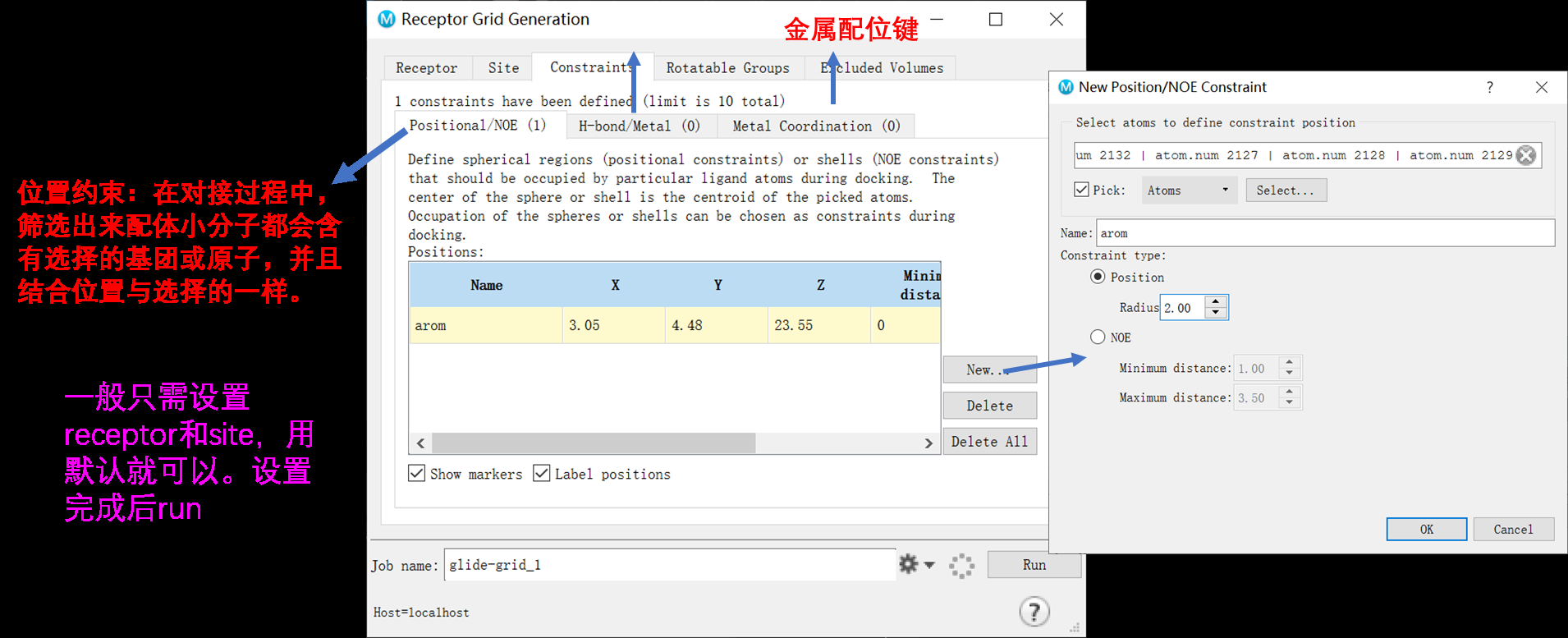
基于实验数据、结构分析,认为一些相互作用对受体-配体相互作用很重要。把这些相互作用设为约束条件,在对接运算早期就排除不符合条件的相互作用。
3.对接与虚筛
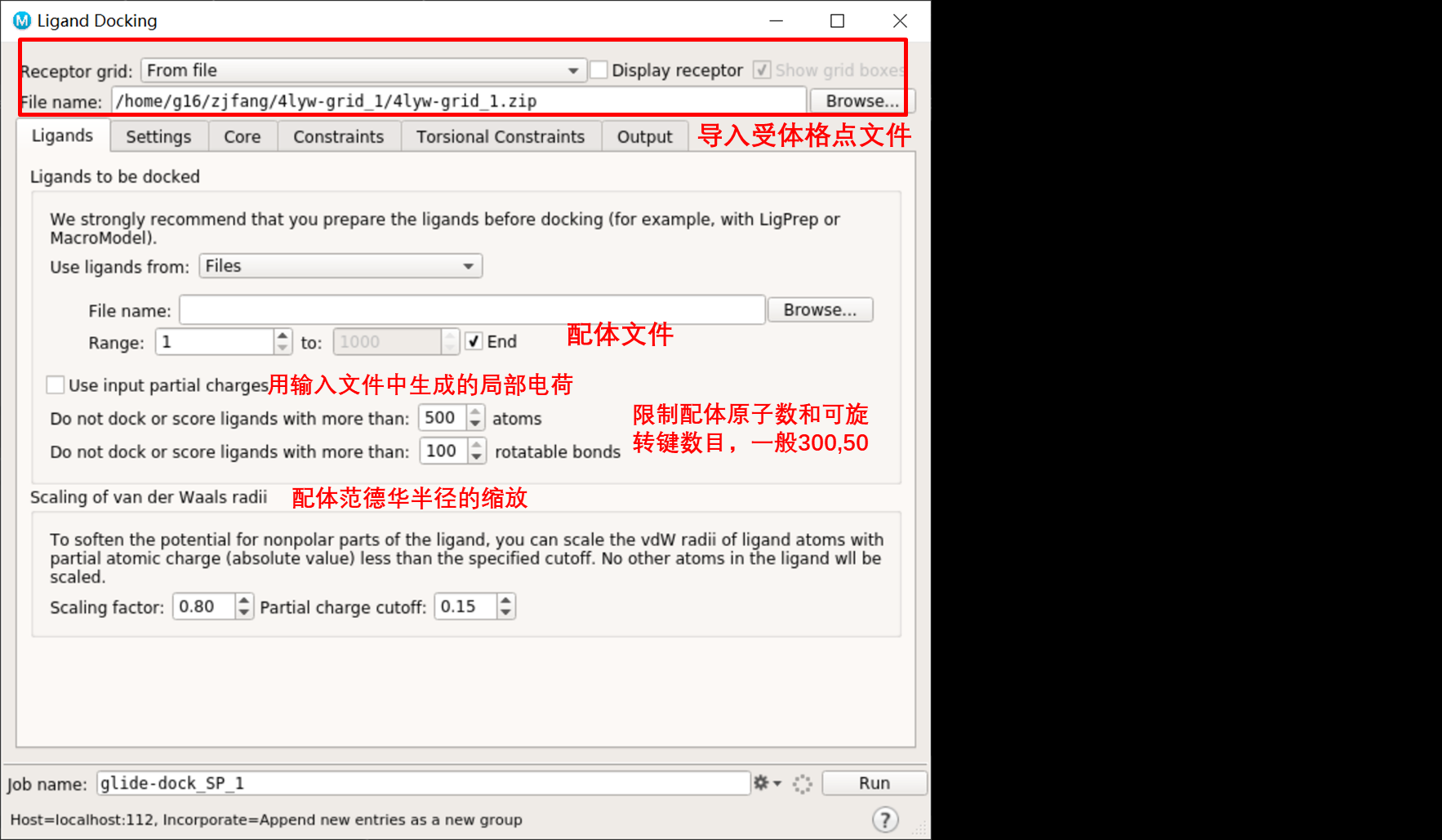
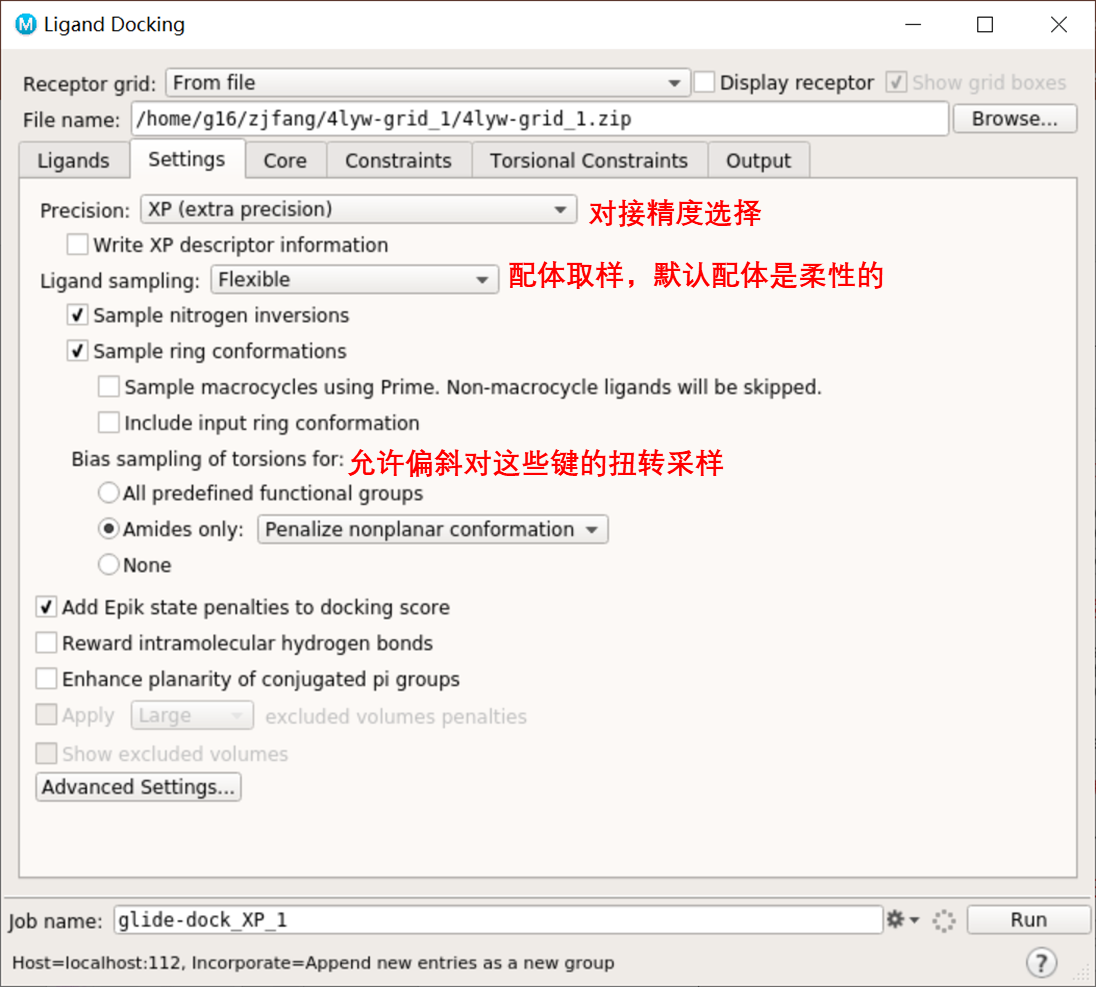
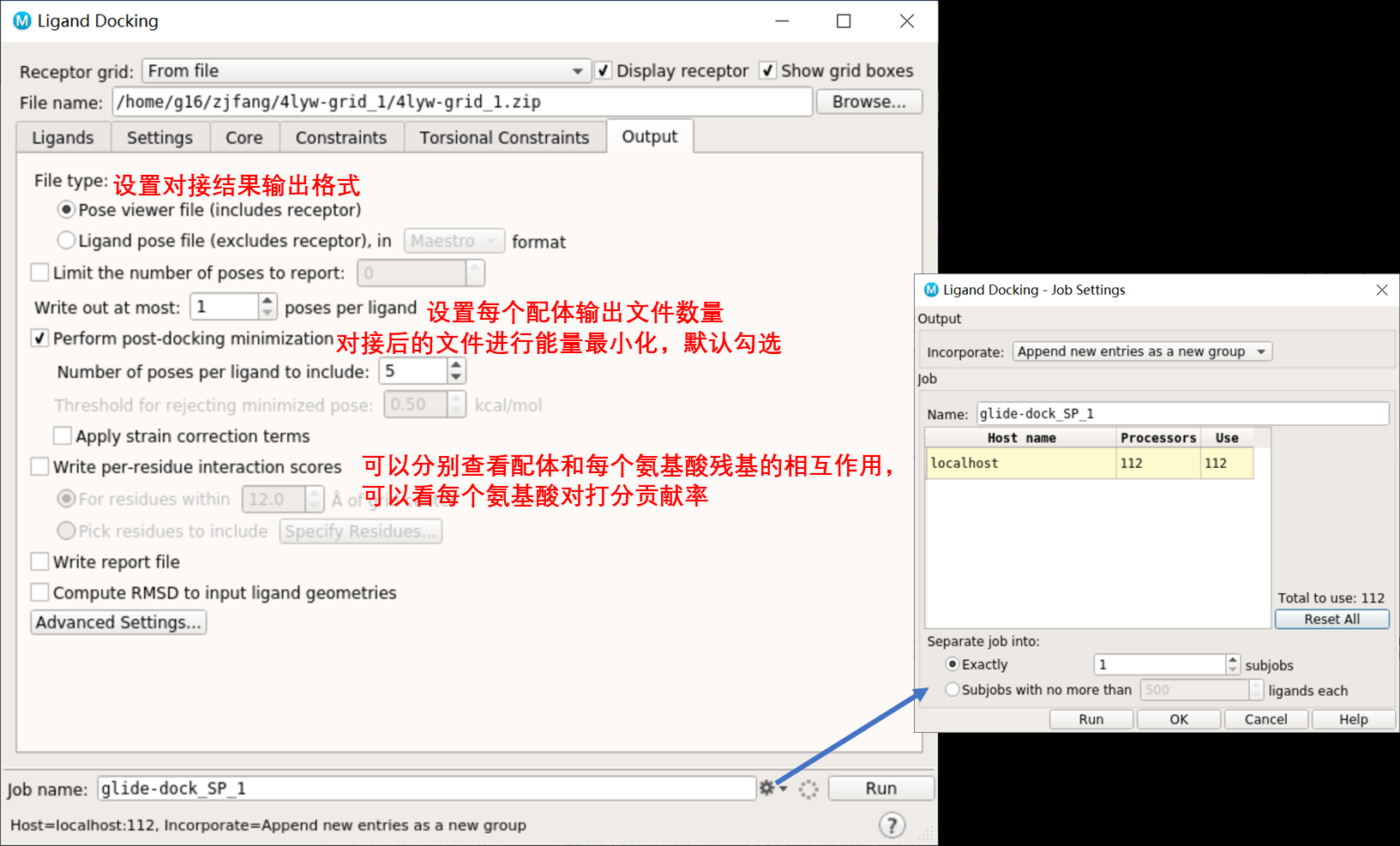
4.后处理
4.1复现结果评价
主要考察对接的配体是否符合与晶体结构一致。导出对接结果,pymol打开,观察重叠的好不好,也可以使用下面的脚本计算小分子之间的RMSD: rmsd_2_mol.py pymol file-run script 选择脚本所在位置,选中参考分子,对接分子分别为 obj01 obj02 rmsd_three obj01 obj02 即可计算出RMSD,2以内都还行,算的不准,做个参考即可,主要是观察。
4.2数据处理
如果分成多个ligand文件进行的,首先将所有HTVS文件夹下的所有pv.maegz(结果文件)复制到新的文件夹下(当然也可以不复制)
- cp -r glide-dock_HTVS*/*pv.maegz new/
使用glide_merge将文件中所有打分优于-7的结果,输出到新的test文件
- glide_merge HTVS_analysis/* -c -7 -o score-7.maegz
注意:这里的打分是可调整的,一般至少要优于-7。调整打分的目标是使得符合要求的分子数满足接下来的任务需求(提高精度对接/送样……)
接下来使用canvas处理结果(24服务器上没有,23上有,也可以用本地的)
- canvas
然而导入后docking score会变为重原子平均score,用maestro打开,table-右键export to canvas可以避免这个问题。 导入score-7.maegz,会报operation failed的错误(蛋白体系原子数目过多),不需理会,选择yes to all。 加载完成后全选表格所有,右侧选binary fingerprints,右键open,finger type选structural keys,其他不用改,create。选incorparate autamatically,设置cpu数量(跑得不慢,不需要太多核)。 完成后全选表格,右侧选Leader-Follow Clustering,右键open,fingerprint column选刚才生成的(默认是fp_maccs_01),cluster radius改为0.2(即相似度0.8以上的聚为一类),create cluster。这里未必选择这个聚类方法,只是这个方法能选阈值(吴乐云师姐)。 完成后全选,右键export to maestro。 maestro:tables,上方show family-canvas all。上方sort-勾选group entries by property before sorting-options-点击property项旁边的按钮,选择LFClust……:Cluster-ok。sort entries项点击add下,选docking score-sort all。 最上方select-select from groups-选择 entries in each group(每组选打分第一的)-select,导出保存maegz文件
4.3结果整理
打开2017版的maestro,打开所保存的文件,tables,全选,表格上的title,右键clear,最上方property-merge
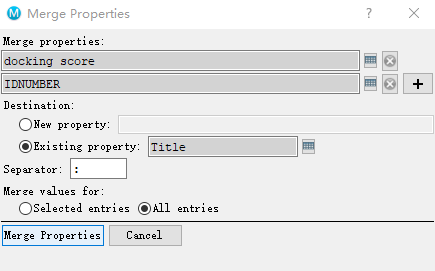
导出为maegz文件,pymol打开,设置一下显示格式,最后保存为pse文件,方便看结构。可以自己先看一遍,把明显不合理的结果去除。 右键export-export to canvas,上方tasks-properties-calculate-选择分子量,重原子计数,其他都不需要。incoprat automatically,计算。 完成后右键全选,导出为xlsx,选择需要导出的性质(一般需要包括分子量,logP,logS,IDNUMBER,score,重原子数)即可。 注意:仅有2017版本能导出带有结构的excel文件,需要win上安装这个版本的。目前在22224:/home/lywu/lywu/software有安装包。
注意:4.2和4.3中使用maestro和canvas处理数据,但是经常会有bug,建议每步检查一下docking score,有时会出现导入导出问题,需要灵活应对,在服务器2018版和本地2017版,maestro和canvas中灵活export。
5.参考
本教程参考,引用了来自张建芳师姐,韩家鑫师兄的教程,同时,周兆寅师弟对原有方法做出了更新,并提供了大量实际指导。