
配体准备
- # 22224
- # 172.21.85.24
- # 使用薛定谔优化结构(图形化界面操作)
- # pH = 8.0,手性从3D结构定(晶体结构),力场为2017版的默认力场OPLS3
- conda activate ambertools
- # 高斯优化
- # 注意检查体系的电荷和自选多重度(eg.将T3的gjf电荷0 1改为1 1)
- antechamber -i lig.sdf -fi sdf -o lig.gjf -fo gcrt -at gaff2 -gn "%nproc=4" -gm "%mem=4GB" -gk "#B3LYP/6-31G* em=gd3bj pop=MK iop(6/33=2,6/42=6) opt" -rn MOL -nc 1
- -nc后面加电荷数,电荷为1就是1,电荷为-1就是-1,在gif文件里有体现
- 自旋多重度计算:(未配对的正电子数-未配对的负电子数)*2+1=1 一般都为1,遇到金属离子需要特别注意
- gif文件如下
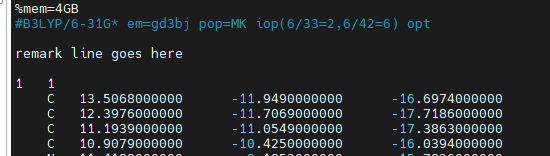
- 第一个1指的是电荷数,电荷为-1就是-1
- 第二个1指的是自旋多重度
- #把gjf文件转成log文件,转换之前检查电荷和自旋多重度
- g16 lig.gjf
- nohup g16 lig.gjf &
- # 基于高斯优化日志文件,拟合RESP电荷,生成AMBER需要的参数文件(prep)
- antechamber -i lig.log -fi gout -o lig.prep -fo prepi -c resp;parmchk2 -i lig.prep -f prepi -o lig.frcmod -a y
- #这里会生成很多文件,包括NEWPDB.PDB
- 下面将进行两次对齐!
- 这一步是为了后面将坐标信息对其。注意:必须以高斯产生的小分子sybmol为基准,因为之前产生的键连信息lig.prep是高斯坐标为基准产生的。但我们需要的是对接的原子坐标信息,所以两次对齐的基准不一。第一次对齐(symbol):高斯产生的小分子为基准(NEWPDB.PDB)。第二次对齐(coordinate):对接的晶体结构为基准(crtstal.pdb)为基准。
- #删除H,获取坐标信息sdf
- gv里分别打开 NEWPDB.PDB 与 crystal.sdf(晶体结构配体文件):打开GV tools-atom list--鼠标点击symbol列-rows-sort selected -拖动鼠标选择所有H-edit-delete-selected atoms),接着仍在atom list中,edit-z matrix-standardize,分别保存。得到 gv-NEWPDB.pdb gv-crystal.pdb
- #第一次对齐
- GV里 检查 gv-NEWPDB.pdb gv-crystal.pdb,更改gv-crystal.pdb (atom list - Tag),使其Tag与gv-NEWPDB.pdb 完全一致。
- #第二次对齐
- dos2unix *
- grep -v "CONECT" gv-NEWPDB.pdb |cut -b 1-26 > symbol
- cat gv-crystal.pdb | cut -b 29-78|sed '1,2d'|sed '1i Combineing the coordinates from the crystal structure with the atomic numbers from the Gaussian optimized structure'> coordinate
- paste -d " " symbol coordinate|sed '1i TITLE lig.pdb'> lig.pdb
- ### 可视化检查