
(小分子)卤键-EP模型在gromacs中的实现
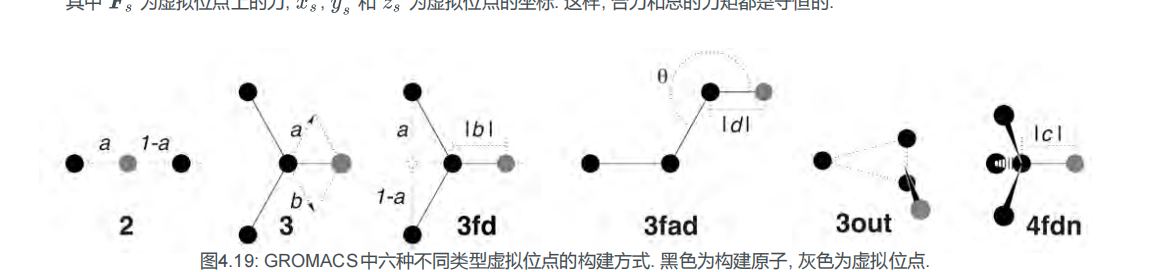
一、小分子处理
1. gauss计算ESP(略)并拟合小分子RESP电荷
计算ESP这一步说明:个人认为若要使用小分子优化结构作为MD的初始小分子结构则在高斯计算的时候就优化,否则就高斯计算的时候就不要优化了(RESP电荷依赖于结构;且后续EP参数拟合的时候要针对高斯计算后的结构进行处理,这样可以避免繁琐的对应操作)
- antechamber -i xxx.log -fi gout -o xxx.mol2 -fo mol2 -c resp -rn xxx -at gaff
2. 拟合带EP的resp电荷
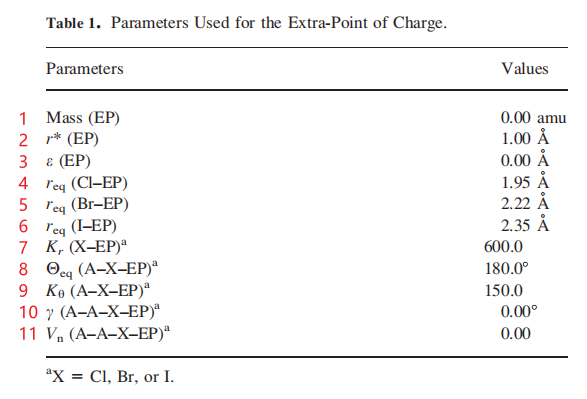
此处的EP距离参数可以使用文献里给的,也可以用自己拟合的,根据需要选:
2.1 生成小分子fchk文件:(要用g16的formchk)
- #GPU4n
- /home/chpeng/software/g16/g16/formchk xxx.chk
2.2 在Multiwfn中计算含EP的RESP电荷:
- xxx.fchk
- 7
- 18
- 4
- 2
- 0
- 0
- 9
- EP_xxxA.txt
- 2
- 1.88 (I)
- 1.75 (Br)
- 1.64 (Cl)
2.3 查看Multiwfn最后输出结果(注意RMSE和RRMSE),保存RESP输出文件即可
- EP_xxxA.txt的书写格式说明:
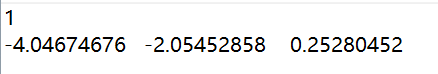
第一行1表示有一个点,第二行为EP的坐标(相对于当前的小分子),EP的坐标可以用GaussView打开小分子文件,添加原子调整合适的距离和角度之后查看原子坐标即可。
- 使用自己拟合的距离参数——在EP_xxxA.txt文件中把不同距离的EP坐标换上,观察最后生成的RESP电荷对应的RMSE和RRMSE最小值对应的EP距离即为最优EP距离。
- 使用文献中的EP距离参数
文献一(这个参数可能过时了,跑不起来):

文献二(可用):
3. 小分子力场文件、结构(电荷)文件准备
3.1 对(一、1.)中生成的mol2文件进行修改(复制一份)
- 添加EP的ATOM信息

- 替换电荷为添加EP后拟合的小分子RESP电荷
- 添加EP的BOND信息(连接的原子号要注意对应上)
3.2 生成带EP的小分子力场参数
- parmchk2 -i 9H2.mol2 -f mol2 -o 9H2.frcmod -a y
frcmod文件说明:
- MASS
- ep 0.000(1) 0.000 ATTN, no polarizability parameter
- BOND
- i-ep 600.00(7) 0.000(456) ATTN, need revision
- ANGLE
- ca-i-ep 150.000(9) 180.000(8) ATTN, need revision
- DIHE
- ca-ca-i-ep 1 0.000 0.000(10) 0.000 ATTN, need revision
- NONBOND
- ep 1.0000(2) 0.0000 ATTN, need revision
序号对应的位置参数要改一下:
二、蛋白处理
normal
三、复合物拓扑文件生成
tleap
- source oldff/leaprc.ff14SB
- source leaprc.gaff
- loadAmberParams frcmod.ions234lm_1264_tip3p
- loadAmberParams frcmod.ionsjc_tip3p
- set default PBradii mbondi2
- loadamberparams 9H2.frcmod
- lig=loadmol2 9H2.mol2
- pro=loadpdb 5o1b_A_addH.pdb
- com=combine {pro lig}
- saveamberparm lig lig.prmtop lig.inpcrd
- saveamberparm pro pro.prmtop pro.inpcrd
- saveamberparm com native.prmtop native.inpcrd
- savepdb com native.pdb
- solvatebox com TIP3PBOX 10
- charge com
- addionsrand com Cl- 5
- charge com
- saveamberparm com complex.prmtop complex.inpcrd
- savepdb com complex.pdb
- quit
- 用acpype转成gmx支持的格式
- acpype -p complex.prmtop -x complex.inpcrd -d
生成complex_GMX.gro/complex_GMX.top
修改complex_GMX.top文件
- 在 [atoms] 和 [bonds] name lists 中间添加 [ virtual_sites2 ]信息
具体自己查gromacs手册,注意最后一列值的换算方式 http://www.mdtutorials.com/gmx/vsites/02_topology.html
- [ virtual_sites2 ]
- 5026 5006 4981 1 -0.8302220
一共有五列,意义分别如下:
EP原子序号(EP) 卤素原子序号(X) 连接卤素原子的序号(C) 1 -(X...EP)/(X...C)
- -(X...EP)/(X...C)解释:
- 若I...EP选择1.73,I...C为2.08378,
- -1.73/2.08378=-0.8302220
- 再修改at_type列的值为Na+/Cl-,否则报错ERROR 1 [file complex_GMX.top, line 11579]: Atomtype NA+ not found

四、跑gromacs
(涉及的mdp输入文件可以问组里跑MD的同学要)
1.能量最小化
- gmx_mpi grompp -f em.mdp -p complex_GMX.top -c complex_GMX.gro -r complex_GMX.gro -o em.tpr -maxwarn 1000
- nohup gmx_mpi mdrun -v -deffnm em &
或:最小化用CPU跑比较快(用ntomp跑CPU 记得设环境变量为一样的)
- gmx_mpi grompp -f em.mdp -p lig_GMX.top -c lig_GMX.gro -r lig_GMX.gro -o em.tpr -maxwarn 1000
- export OMP_NUM_THREADS=24
- gmx_mpi mdrun -ntomp 24 -v -deffnm em
2.NVT
- gmx_mpi grompp -f nvt.mdp -p complex_GMX.top -c em.gro -r em.gro -o nvt.tpr -maxwarn 1000
- nohup gmx_mpi mdrun -v -deffnm nvt &
3.NPT
- gmx_mpi grompp -f npt.mdp -p complex_GMX.top -c em.gro -r em.gro -o npt.tpr -maxwarn 1000
- nohup gmx_mpi mdrun -v -deffnm npt &
4.MD
- gmx_mpi grompp -f md.mdp -p complex_GMX.top -c npt.gro -r npt.gro -o md.tpr -maxwarn 1000
- nohup gmx_mpi mdrun -v -deffnm md &
分析:和常规分析一样