
算熵方法总结
- MMPBSA.py 算熵变
熵变计算有两种方法,QH和NMA(NMA比较常见)
命令参数和计算焓变是一样的,in文件要单独设置文件
NMA示例文件:
- Input file for running PB and GB in serial
- &nmode
- nmstartframe=1, nmendframe=100,
- nminterval=30, nmode_igb=5, nmode_istrng=0.1,
- /
QH示例文件
- Input file for running PB and GB
- &general
- startframe=1, endframe=5, interval=1,entropy=1, keep_files=0,
- /
执行命令:
- mpirun -np 10 MMPBSA.py.MPI -O -i qh.in -o qh_entropy.dat -cp ../native.prmtop -rp ../protein.prmtop -lp ../ligand.prmtop -y test-fit.pdb
PS:QH在计算的时候会占用大量的RAM(运行内存),所以在提交的时候要注意只提交10个及以下的核数,不同的体系大小,不同的服务器情况不一样,需要测试,注意观察nohup out文件,没有报错才能正常算,即使任务还在运行,有报错也算不出来。经测试,八千个原子的体系,GPU4N可以交10个核,普通服务器交5个可以正常运行,并且交5个和10个的速度差不多。
帧数太多也会报错,猜测是运行内存不足
NMA的计算不支持charmm力场
- gmx_MMPBSA
221服务器上安装的是1.4.3版本
因为需要的ambertools版本较高,所以安装的时候是在amber_20的环境下安装的
安装之前,环境变量里的amber版本一定要改成20
- ./amber20_src/miniconda/bin/python3 -m pip install gmx-mmpbsa
- vi ~/.bashrc
- export PATH="/path_to_amber_install/amber20/miniconda/bin:$PATH" 加入环境变量
- source ~/.bashrc
由于该版本里面的mpi与gmx_mpi不兼容,而脚本运行时需要用到gmx,所以需要重新编译一个非mpi的gmx并且加入环境变量,便于脚本运行时查找
MPI运行示例
- mpirun -np 10 gmx_MMPBSA MPI -O -i ie.in -ci index.ndx -cg 10 11 -cp native_noglycan.top -ct md-noglycan-100-400.pdb -cs look.pdb
IE脚本算熵变
用的是华东师范大学张增辉老师组开发的IE算法脚本(脚本在221服务器上 /home/jawang/software/IE)
参考文献:Interaction entropy for protein-protein binding
- Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein-Ligand Binding Free Energy
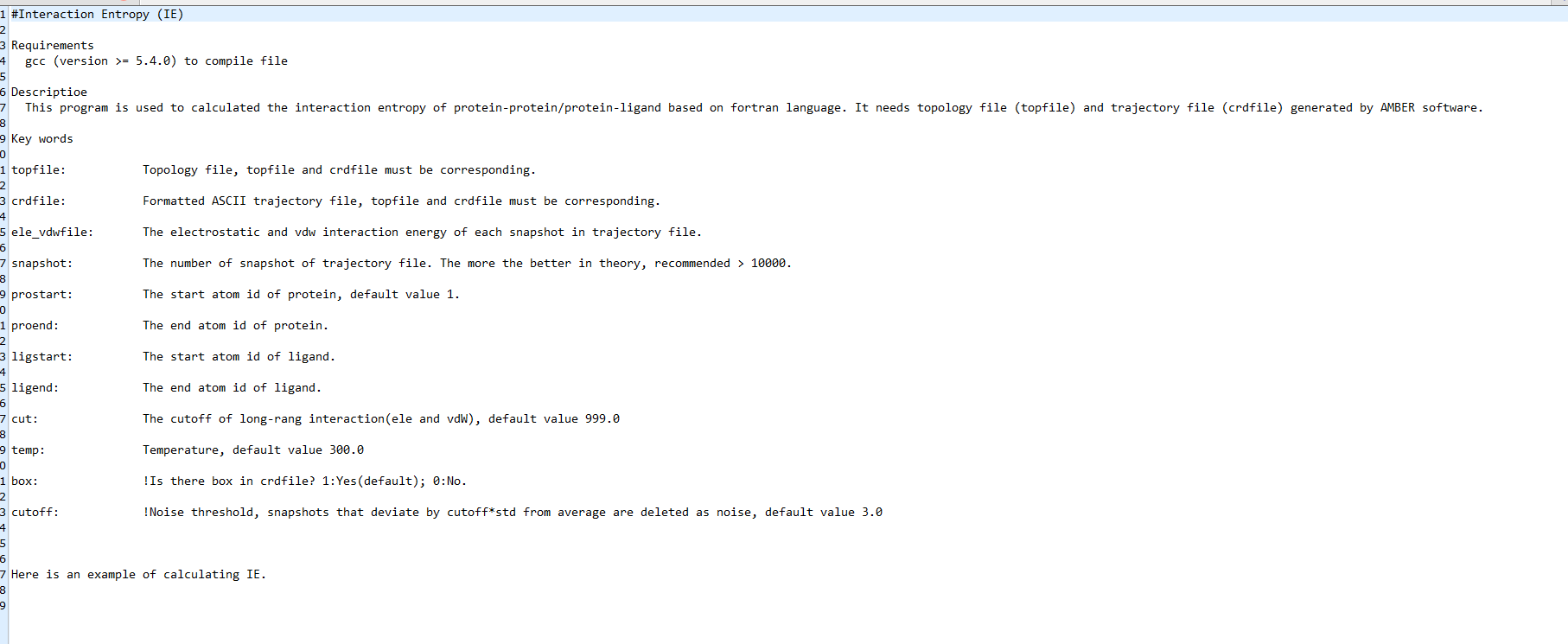
修改run.sh中的相关参数即可运行
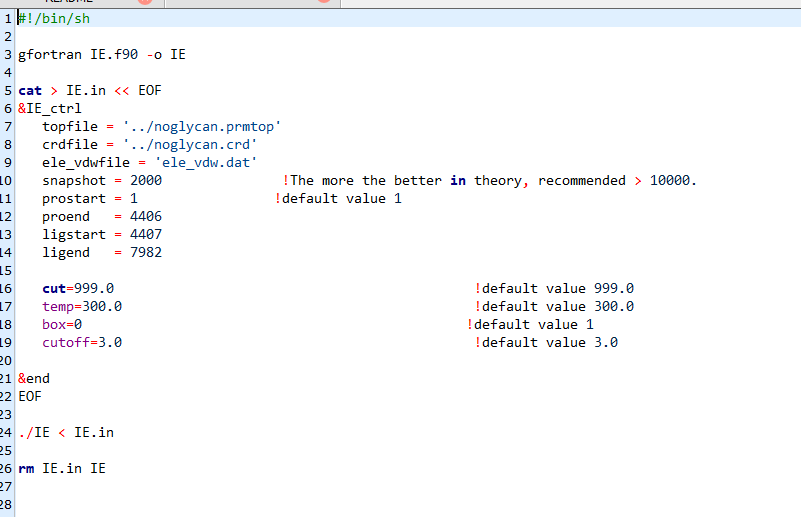
PS: 文件名最好不要出现下划线等符号,不然会报错,另外charmm力场转成的amber参数与tleap生成的格式有些不一样,比如电荷前者是一排3个,后者是5个,所以需要改代码,改好的脚本在IEchamber.f90中,改之前的是IE.f90,运行时在run.sh中切换两个脚本即可。
- Conformational ENtropy CALCulations
- CENCALC
221服务器上有,但是要结合状态和非结合状态单独算,不能算复合物