
amber TI 热力学积分计算结合自由能
理论:
研究系统的两个状态的自由能变化也由一个耦合变量λ连接, 初态时λ= 0,终态时λ= 1. 通过连续改变λ, 得到一系列介于初态和终态的中间态, 通过对这些中间态的取样, 最终计算得到自由能变化数据. 以 H 表示研究体系在某个状态λ下的能量, 则其具体的自由能计算公式为:
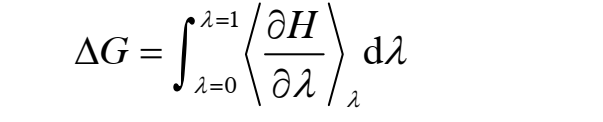
由于在实际应用中, 无法得到连续变化的相关数据, 只能为λ设定一些差异较小的离散值. 通过对研究体系在不同λ对应的状态下的多次取样, 估计出研究体系的能量以后, 由下面的自由能计算公式, 就可得到相关的自由能变化数据:
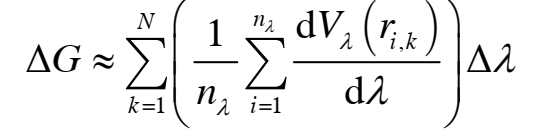
其中 N 为计算过程中涉及到的研究体系状态的个数,也即λ的个数; nλ为对研究体系的某个状态的取样次数; Vλ则是研究体系在状态λ下的势能; 坐标 ri, k 则与研究体系在状态lamda下的玻尔兹曼概率相关, 其中下标i 和 k 分别指对研究体系每个状态的取样数以及涉及到的研究体系状态的总个数.
A general assumption in all TI calculations is that the system does not undergo significant conformational changes during the transformation, otherwise MD simulations will most likely not sample enough phase space for converged results. Other free energy techniques can be used if free energies from conformational changes need to be computed.
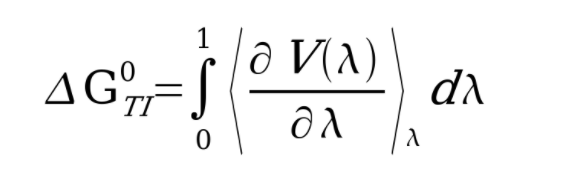
在不同的λ下平行模拟,最后根据得出的能量进行积分运算
计算不同配体与同一个受体的结合自由能差值
步骤:
1. Setting up the vdW+bonded transformation step
不同的配体放在同一个PDB文件当中,主干的坐标一致,残基名称不同,tleap会根据残基的名称加上缺失的原子
tleap处理
tleap=$AMBERHOME/bin/tleap
basedir=leap
$tleap -f - <<_EOF
# load the AMBER force fields
source oldff/leaprc.ff14SB
source leaprc.gaff
loadAmberParams frcmod.ionsjc_tip3p
# load force field parameters for BNZ and PHN
loadoff $basedir/benz.lib
loadoff $basedir/phen.lib
# load the coordinates and create the complex
# 配体的pdb文件中只有两个配体相同的部分,只有名称和编号不同,tleap会根据lib文件和名称补上残缺的原子
ligands = loadpdb $basedir/bnz_phn.pdb
complex = loadpdb $basedir/181L_mod.pdb
complex = combine {ligands complex}
# create ligands in solution for vdw+bonded transformation
solvatebox ligands TIP3PBOX 12.0 0.75
addions ligands Cl- 0
savepdb ligands ligands_vdw_bonded.pdb
saveamberparm ligands ligands_vdw_bonded.parm7 ligands_vdw_bonded.rst7
# create complex in solution for vdw+bonded transformation
solvatebox complex TIP3PBOX 12.0 0.75
addions complex Cl- 0
savepdb complex complex_vdw_bonded.pdb
saveamberparm complex complex_vdw_bonded.parm7 complex_vdw_bonded.rst7
quit
2. Running MD to provide sane starting coordinates
正常步骤能量最小化、升温、加压(复合物、配体都要进行)
in文件中有特殊的参数设置需要仔细看一下?
minimisation
&cntrl
imin = 1, ntmin = 2,
maxcyc = 100,
ntpr = 20, ntwe = 20,
ntb = 1,
ntr = 1, restraint_wt = 5.00,
restraintmask='!:WAT & !@H=',
icfe = 1, ifsc = 1, clambda = 0.5, scalpha = 0.5, scbeta = 12.0,
#icfe 启动自由能计算
#ifsc 0 SC potentials are not used (default) 1 SC potentials are used. Be sure to use prmtop files that are suitable for this, i.e. not-containingdummy atoms
logdvdl = 0,
#
timask1 = ':BNZ', timask2 = ':PHN',
scmask1 = ':BNZ@H6', scmask2 = ':PHN@O1,H6'
/
&ewald
/
heat,press的in文件都差别不大
3. Extracting the Coordinates from the MD simulation
将加压之后的复合物、配体press.rst7分成三部分---单独的两个配体以及溶剂部分
4. Setting up the Decharging and Recharging steps
用上面VDW+bond 经过加压之后的结构和水盒子,是为了保证charge,vdw,bond的起始坐标是一模一样的
用tleap生成ligand和complex的decharge和recharge的参数文件,decharge的参数文件,两个ligand1在一个文件中,recharge的参数文件,两个ligand2在同一个文件中
5.生成in文件,做自己的体系的时候需要把
6.RUN TI
heat之后正式跑TI,decharge, recharge,vdw和bond都要跑N个window
7.分析数据
python脚本要用python2运行,analyse.sh
side-chain mutation
基本操作与上述ligand是一样的
1.先准备参数文件,WT和mut要放在同一个文件中,先将wt_mut组成的ligands, complex加水,加离子,生成prmtop和rst7文件之后,用parmed merge处理,接着能量最小化、升温加压之后,再用cpptraj将水-离子和蛋白、配体分开成为三部分,再用tleap处理乘胜recharge和decharge的文件,parmed合并