
Alchemical_analysis 算蛋白-小分子deltaG
208服务器上有
gromacs 算蛋白配体绝对结合自由能
大致的流程分为三部分:
- 算配体的decoulping过程,算溶剂化自由能
- 算复合物的decoulping过程(处理数据时取负号)
- 限制势的矫正项
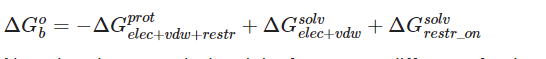
原理:
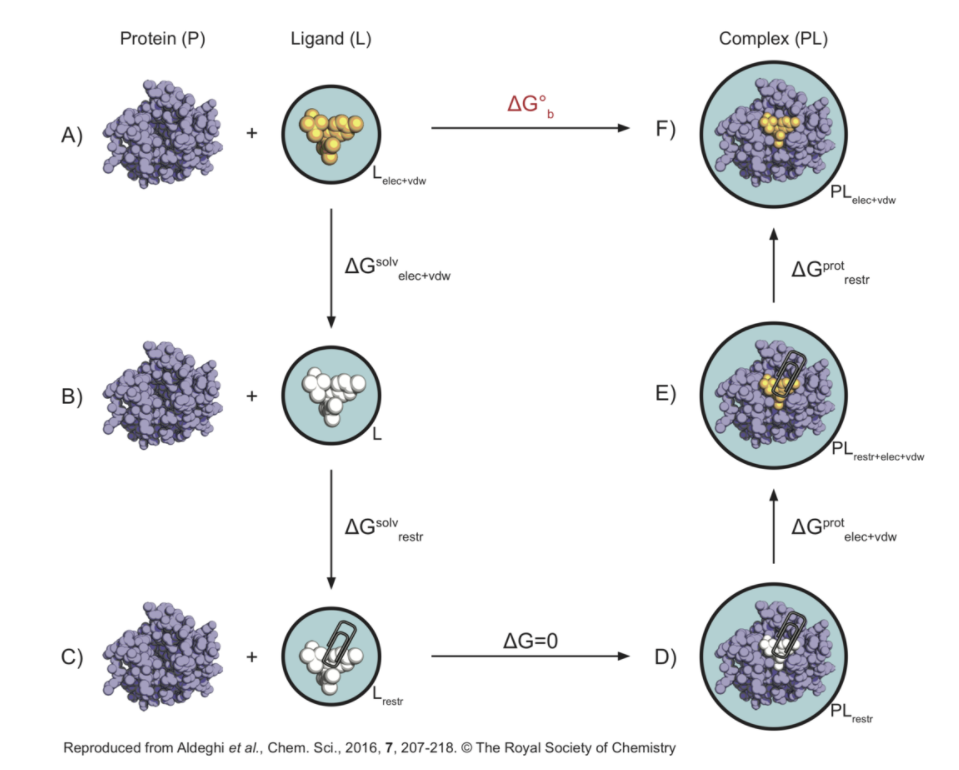
在这个热力学循环中,我们需要模拟的系统是由一个黑色的圈表示,回形针表示限制的存在,白色配体意味着它不是与环境交互(即解耦)和浅蓝色背景明确水的存在。
右边包括对蛋白质配体复合物的模拟,而左边包括对溶液中配体的模拟。
我们要做的就是从左上角这个解离状态A通过一系列非物质(或炼金术)中间状态(像B, C, D, E)到达右上角表示物理束缚态F状态,左上角的周期(状态)。
A-B:从溶液中解耦
B-C:加入限制,为了使其位置和方向接近结合状态
C-D:结合到口袋中,但与外界环境的作用力仍处于关闭状态
D-E:作用力被逐步打开
E-F:去除限制
准备参数文件
ligand:
- 高斯生成参数文件
从复合物文件中分离出小分子的PDB文件,修改其中的原子名称,原子名称依次为N1 N2 C1 C2 H1 H2往下,配体名称为LIG
生成小分子的参数文件:
- antechamber -i TSG12.pdb -fi pdb -o TSG12.gjf -fo gcrt
修改gjf文件
%chk= TSG12
%nproc=16
#B3LYP/6-31G* SCF=tight
SCRF=SMD Test Pop=MK iop(6/33=2) iop(6/42=6) opt
注意不要有多余的空行,不然会报错,多重自旋度一般为1,如果小分子带电荷,要自己手动修改其中的参数。
- g09 TSG12.gjf
(关掉界面的话,高斯会自己断掉,可以用exit退出)
- antechamber -i TSG12.log -fi gout -o ligand_TSG12.prep -fo prepi -c resp -nc 净电荷
- parmchk -i ligand_TSG12.prep -f prepi -o ligand_TSG12.frcmod -a y
将prep文件中的残基名称改为对应的小分子名称(LIG)
- tleap生成配体参数文件
- source oldff/leaprc.ff14SB
- source leaprc.gaff
- loadAmberParams frcmod.ions234lm_1264_tip3p
- loadAmberParams frcmod.ionsjc_tip3p
- loadamberparams ligand_TSG12.frcmod
- loadamberprep ligand_TSG12.prep
- lig=loadpdb TSG12.pdb
- saveamberparm lig native.prmtop native.inpcrd
- savepdb lig native.pdb
- solvatebox lig TIP3PBOX 10
- charge lig
- saveamberparm lig ligand.prmtop ligand.inpcrd
- savepdb lig ligand.pdb
- quit
- python的parmed包转成gromacs的参数文件
- import parmed as pmd
- amber=pmd.load_file("ligand.prmtop","ligand.inpcrd")
- amber.save("ligand.gro")
- amber.save("ligand.top")
- 修改gro,top文件
vi ligand.gro
:1,$s/WAT/SOL/g
:1,$s/Na+/NA /g
:1,$s/Cl-/CL /g
vi ligand.top
:1,$s/WAT/SOL/g
:1,$s/Na+/NA /g #atomtype那里钠离子改成Na(top文件最前面和后面的离子信息部分)
:1,$s/Cl-/CL /g #atomtype那里氯离子改成Cl
:1,$s/LIG /ligand/g #atomtype那里钠离子改成Na
:1,$s/Generic title/ligand /g
- 生成位置限制文件,并在top文件适当的地方加入
; Ligand position restraints
#ifdef POSRES
#include "posre_ligand.itp"
#endif
- gmx_mpi genrestr -f ligand.gro -o posre_ligand.itp
- 根据不同的lambda值生成MDP文件,跑配体单独的MD
这一步用的教程中的MDP参数,如有需要可以改
nohup ./run.sh &
- 将dh_dl文件总结到新的文件夹中
./dh_dl.sh
dh_dl.sh的内容:
- if [ ! -d "dHdl_files" ]; then
- mkdir dHdl_files
- fi
- for i in $(ls | grep "lambda.*"); do
- lam="${i##*.}" #这表示在i中取以.分割的后半部分内容,即i=lambda.0时,lam=0
- cp ./$i/PROD/dhdl.xvg ./dHdl_files/dhdl.$lam.xvg
- done
- 用alchemical-analysis计算
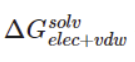
alchemical_analysis -d dHdl_files -t 300 -s 100 -u kcal -w -g #-d dh_dl所在文件夹,-t温度,-s跳过的时间,ps为单位,-u输出单位,-w输出相空间重叠矩阵,-g输出不同方法在相邻窗口算出的能量差别
complex:
- tleap生成参数文件
- source oldff/leaprc.ff14SB
- source leaprc.gaff
- loadAmberParams frcmod.ions234lm_1264_tip3p
- loadAmberParams frcmod.ionsjc_tip3p
- loadamberparams ligand_TSG12.frcmod
- loadamberprep ligand_TSG12.prep
- pro=loadpdb tg2_pock4_model_noH.pdb
- saveamberparm pro native.prmtop native.inpcrd
- savepdb pro native.pdb
- charge pro
- addions pro Cl- 2
- solvatebox pro TIP3PBOX 10
- saveamberparm pro complex.prmtop complex.inpcrd
- savepdb pro complex.pdb
- quit
- 修改gro,top文件
top文件中需要解耦的部分(也就是mdp文件中couple-moltype对应的部分需要单独成为moleculartype,并且名字相对应,可以将配体单独生成ligand.itp文件后,在适当的位置加入 #include "ligand.itp")
vi complex.gro
:1,$s/WAT/SOL/g
:1,$s/Na+/NA /g
:1,$s/Cl-/CL /g
vi complex.top
:1,$s/WAT/SOL/g
:1,$s/Na+/NA /g #atomtype那里钠离子改成Na
:1,$s/Cl-/CL /g #atomtype那里氯离子改成Cl
:1,$s/LIG /ligand/g
:1,$s/system1/Protein/g
:1,$s/Generic title/Complex /g
- 生成位置限制文件,并在top文件适当的地方加入
- gmx_mpi genrestr -f complex.gro -o posre_ligand.itp
- gmx_mpi genrestr -f complex.gro -o posre_protein.itp
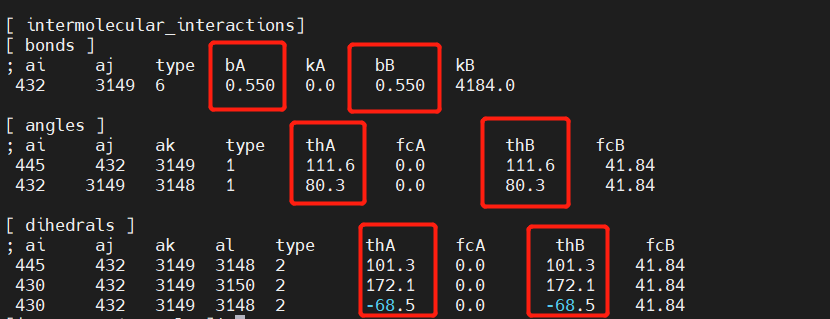
- 根据gro文件,选配体刚性环中的三个原子以及附近相互作用的残基主链上的三个原子,用pymol测量距离,角度,二面角信息加入参数文件中作为限制
其余的参数参考的是example
- 根据不同的lambda值生成MDP文件,跑复合物的MD
这一步用的教程中的MDP参数,如有需要可以改
nohup ./run.sh &
- 将dh_dl文件总结到新的文件夹中
./dh_dl.sh
- 用alchemical-analysis计算
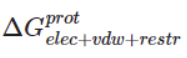
alchemical_analysis -d dHdl_files -t 300 -s 100 -u kcal -w -g #-d dh_dl所在文件夹,-t温度,-s跳过的时间,ps为单位,-u输出单位,-w输出相空间重叠矩阵,-g输出不同方法在相邻窗口算出的能量差别
- 计算矫正项
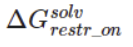

根据实际情况更改restraints.py文件中的相关变量 T,r0,thA,thB,K_*
python restraints.py #运行结果取dG_on
restraints.py的内容
- import math
- import sys
- #===================================================================================================
- # INPUTS
- #===================================================================================================
- K = 8.314472*0.001 # Gas constant in kJ/mol/K
- V = 1.66 # standard volume in nm^3
- T = 300.0 # Temperature in Kelvin
- r0 = 0.654 # Distance in nm
- thA = 88.8 # Angle in degrees
- thB = 32.9 # Angle in degrees
- K_r = 4184.0 # force constant for distance (kJ/mol/nm^2)
- K_thA = 41.84 # force constant for angle (kJ/mol/rad^2)
- K_thB = 41.84 # force constant for angle (kJ/mol/rad^2)
- K_phiA = 41.84 # force constant for dihedral (kJ/mol/rad^2)
- K_phiB = 41.84 # force constant for dihedral (kJ/mol/rad^2)
- K_phiC = 41.84 # force constant for dihedral (kJ/mol/rad^2)
- #===================================================================================================
- # BORESCH FORMULA
- #===================================================================================================
- thA = math.radians(thA) # convert angle from degrees to radians --> math.sin() wants radians
- thB = math.radians(thB) # convert angle from degrees to radians --> math.sin() wants radians
- arg =(
- (8.0 * math.pi**2.0 * V) / (r0**2.0 * math.sin(thA) * math.sin(thB))
- *
- (
- ( (K_r * K_thA * K_thB * K_phiA * K_phiB * K_phiC)**0.5 ) / ( (2.0 * math.pi * K * T)**(3.0) )
- )
- )
- dG = - K * T * math.log(arg)
- print "dG_off = %8.3f kcal/mol" %(dG/4.184)
- print "dG_on = %8.3f kcal/mol" %(-dG/4.184)
- 最后的delta_G计算
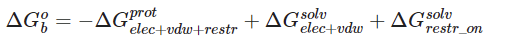
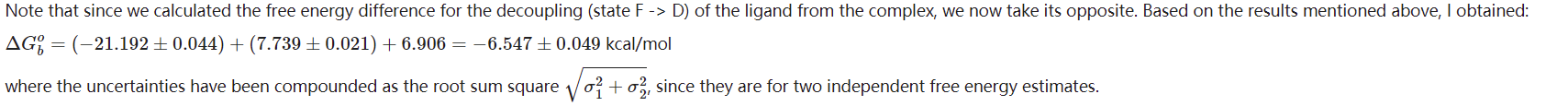
- 配体MDP 文件的重新生成:
根据需求更改模板mdp文件的参数以及py脚本中lambda list的设置,在MDP文件夹中执行
python mk_mdp_ligand.py #要用python3以上版本
mk_mdp_ligand.py的内容
- import os
- import sys
- coul_lambda_list = [0.0,0.2,0.3,0.4,0.5,0.6,0.7,0.8,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0,1.0] #设置库仑力的lambda值
- coul_lambda_nums = len(coul_lambda_list)
- coul_lambda_list2 = [str(i) for i in coul_lambda_list] #转成字符串,以便后续操作
- vdw_lambda_list = [0.0,0.00,0.00,0.00,0.00,0.00,0.00,0.00,0.00,0.05,0.1,0.2,0.3,0.4,0.5,0.6,0.65,0.7,0.75,0.8,0.85,0.9,0.95,1.0] #设置范德华力的lambda值
- vdw_lambda_nums = len(vdw_lambda_list)
- vdw_lambda_list2 = [str(i) for i in vdw_lambda_list] #转成字符串,以便后续操作
- coul_lambda = " ".join(coul_lambda_list2) #生成长字符串,用空格连接
- vdw_lambda = " ".join(vdw_lambda_list2)
- if coul_lambda_nums != vdw_lambda_nums:
- print("coul_lambda_nums" + "不等于" + "vdw_lambda_nums")
- sys.exit() #如果库仑力和范德华的lambda值设置不一致,则退出程序
- else:
- lambda_nums = coul_lambda_nums
- for j in ["ENMIN","NVT","NPT","PROD"]:
- os.system('mkdir ' + j) #创建文件夹
- for i in range(lambda_nums):
- i = str(i)
- file_tem = j.lower() + ".tem.mdp" #模板mdp文件的名称
- file_name = j.lower() + "." + i + ".mdp" #不同lambda对应的mdp文件的名称
- os.system('cp ' + file_tem + ' ' + j + '/' + file_name) #复制模板mdp文件到新的文件夹中
- os.system('sed -i "s/lambda_num/' + i + '/g" ' + j + '/' + file_name) #替换mdp中的lambda_num字符
- os.system('sed -i "s/coul_lambda/' + coul_lambda + '/g" ' + j + '/' + file_name) #替换mdp中的coul_lambda字符
- os.system('sed -i "s/vdw_lambda/' + vdw_lambda + '/g" ' + j + '/' + file_name) #替换mdp中的vdw_lambda字符
以上脚本中特别注意的是os.system的用法,可以在python中调用shell语言,特别注意其中引用变量的写法,单双引号的用法。可以理解成将system()括号中的语句看成是shell中的一条命令,其中字符串与引用变量的连接需要用“str” + variable + "str" 其中的空格也要写上
- 复合物MDP 文件的重新生成:
根据需求更改模板mdp文件的参数以及py脚本中lambda list的设置,在MDP文件夹中执行
python mk_mdp_complex.py #要用python3以上版本
可以注意到complex中多了bonded_lambda的设置,逐步打开限制后,再进行库仑力和范德华的解耦过程
- import os
- import sys
- bonded_lambda_list = [0.0,0.01,0.025,0.05,0.075,0.1,0.2,0.35,0.5,0.75,1.0,1.00,1.0,1.00,1.0,1.00,1.0,1.0,1.0,1.0,1.0,1.0,1.00,1.0,1.00,1.0,1.00,1.0,1.00,1.0]
- bonded_lambda_nums = len(bonded_lambda_list)
- bonded_lambda_list2 = [str(i) for i in bonded_lambda_list]
- coul_lambda_list = [0.0,0.00,0.000,0.00,0.000,0.0,0.0,0.00,0.0,0.00,0.0,0.25,0.5,0.75,1.0,1.00,1.0,1.0,1.0,1.0,1.0,1.0,1.00,1.0,1.00,1.0,1.00,1.0,1.00,1.0]
- coul_lambda_nums = len(coul_lambda_list)
- coul_lambda_list2 = [str(i) for i in coul_lambda_list]
- vdw_lambda_list = [0.0,0.00,0.000,0.00,0.000,0.0,0.0,0.00,0.0,0.00,0.0,0.00,0.0,0.00,0.0,0.05,0.1,0.2,0.3,0.4,0.5,0.6,0.65,0.7,0.75,0.8,0.85,0.9,0.95,1.0]
- vdw_lambda_nums = len(vdw_lambda_list)
- vdw_lambda_list2 = [str(i) for i in vdw_lambda_list]
- coul_lambda = " ".join(coul_lambda_list2)
- vdw_lambda = " ".join(vdw_lambda_list2)
- bonded_lambda = " ".join(bonded_lambda_list2)
- if coul_lambda_nums != vdw_lambda_nums or coul_lambda_nums != bonded_lambda_nums:
- print("coul_lambda_nums,vdw_lambda_nums,bonded_lambda_nums 三者不相等")
- sys.exit()
- else:
- lambda_nums = coul_lambda_nums
- for j in ["ENMIN","NVT","NPT","PROD"]:
- os.system('mkdir ' + j)
- for i in range(lambda_nums):
- i = str(i)
- file_tem = j.lower() + ".tem.mdp"
- file_name = j.lower() + "." + i + ".mdp"
- os.system('cp ' + file_tem + ' ' + j + '/' + file_name)
- os.system('sed -i "s/lambda_num/' + i + '/g" ' + j + '/' + file_name)
- os.system('sed -i "s/bonded_lambda/' + bonded_lambda + '/g" ' + j + '/' + file_name)
- os.system('sed -i "s/coul_lambda/' + coul_lambda + '/g" ' + j + '/' + file_name)
- os.system('sed -i "s/vdw_lambda/' + vdw_lambda + '/g" ' + j + '/' + file_name)
模板MDP文件在服务器217和服务器208上
- 官方推荐的检验模拟是否合格的标准:
