
3.17 md补充实验
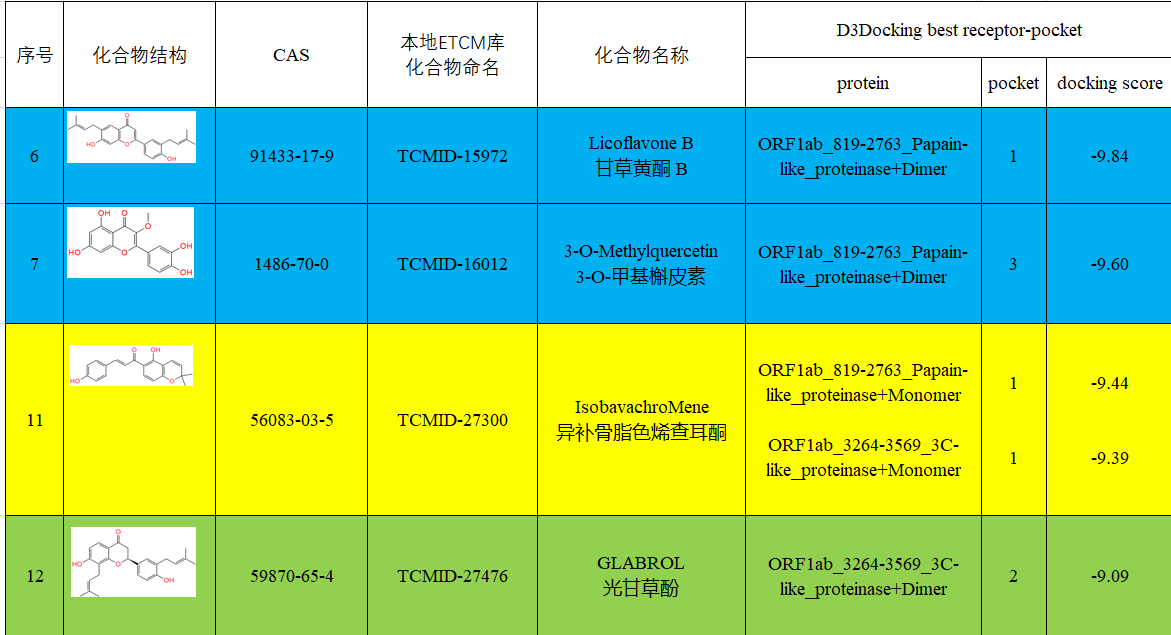
前期工作中,对11个送测活性的成分与mpro(二聚体)/ plpro(三聚体)都做了100ns的CMD模拟。初步分析轨迹,mmgsba计算结合自由能发现,结合自由能与活性数据相关性较差,且涉及PLpro的三聚体的复合物CMD模拟构象波动极大。故计划将CMD模拟部分作为活性测试后的分子机制(蛋白-配体作用)阐明。梳理后的逻辑为:对有活性的4个源自HSBD的关键天然产物进行mpro/plpro 的D3Docking全库对接(所有构象,所有口袋),找打分最好的那个对接结果构建CMD初始构象,跑CMD模拟,进行分析;
打分最好的对接结果(初始构象)
蛋白准备
本次重新准备,全部选用 D3Docking 库中的 受体结构文件(pdb) - pdb2pqr
- 85.23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/pro
- grep -v H0 ../ORF1ab_3264-3569_3C-like_proteinase+Dimer.pdb|grep -v H1
- # 蛋白pdb中原有的H0,H1无法识别,需要全部去掉
- grep -v H0 from_d3dock_pdb/ORF1ab_3264-3569_3C-like_proteinase+Dimer.pdb|grep -v H1 >d3dock_noH_3CL_Dimer.pdb
- grep -v H0 from_d3dock_pdb/ORF1ab_3264-3569_3C-like_proteinase+Monomer.pdb|grep -v H1 >d3dock_noH_3CL_Monomer.pdb
- grep -v H0 from_d3dock_pdb/ORF1ab_819-2763_Papain-like_proteinase+Monomer.pdb|grep -v H1 >d3dock_noH_PL_Monomer.pdb
- grep -v H0 from_d3dock_pdb/ORF1ab_819-2763_Papain-like_proteinase+Dimer.pdb|grep -v H1 >d3dock_noH_PL_Dimer.pdb
- 85.23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/pro/pdb2pqr
- pdb2pqr30 ../d3dock_noH_3CL_Dimer.pdb 3CL_Dimer.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output 3CL_Dimer.pdb
- pdb2pqr30 ../d3dock_noH_3CL_Monomer.pdb 3CL_Monomer.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output 3CL_Monomer.pdb
- pdb2pqr30 ../d3dock_noH_PL_Monomer.pdb PL_Monomer.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output PL_Monomer.pdb
- pdb2pqr30 ../d3dock_noH_PL_Dimer.pdb PL_Dimer.pqr --ff AMBER --ffout AMBER --with-ph 7.4 --pdb-output PL_Dimer.pdb
配体准备
坐标信息(d3dock)
- # 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/ligand/d3dock
- cp ../../../d3dock/results/TCMID-15972/ORF1ab_819-2763_Papain-like_proteinase+Dimer/PDB/TCMID-15972-pocket1-0.sdf 91433-17-9-dock.sdf
- cp ../../../d3dock/results/TCMID-16012/ORF1ab_819-2763_Papain-like_proteinase+Dimer/PDB/TCMID-16012-pocket3-0.sdf 1486-70-0-dock-PL.sdf
- cp ../../../d3dock/results/TCMID-27300/ORF1ab_819-2763_Papain-like_proteinase+Monomer/PDB/TCMID-27300-pocket1-0.sdf 56083-03-5-dock-PL.sdf
- cp ../../../d3dock/results/TCMID-27300/ORF1ab_3264-3569_3C-like_proteinase+Monomer/PDB/TCMID-27300-pocket1-0.sdf 56083-03-5-dock-3CL.sdf
- cp ../../../d3dock/results/TCMID-27476/ORF1ab_3264-3569_3C-like_proteinase+Dimer/PDB/TCMID-27476-pocket2-0.sdf 59870-65-4-dock-3CL.sdf
质子化预测
- 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/ligand/d3dock/ligprep
- for a in
`cat index`;do ~/software/Schrodinger_Suites_2021-2/ligprep -R e -epik -ph 7.4 -pht 0.0 -isd ../${a}.sdf -osd ./${a}.sdf;done
高斯优化
- 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/ligand/g16
- # 使用antechamber生成配体小分子参数(85.23)
- for i in
`cat index`;do mkdir $i;cd $i;antechamber -i ../../d3dock/ligprep/${i}.sdf -fi sdf -o $i.gjf -fo gcrt;sed -i '1,3d' $i.gjf;sed -i '1i %mem=4GB\n%nproc=4\n#B3LYP/6-31G* Pop=MK iop(6/33=2) iop(6/42=6) opt' $i.gjf;cd ../;done - # 提交 高斯优化任务 23:05
- for i in
`cat index`;do cd $i;antechamber -i $i.log -fi gout -o $i.prep -fo prepi -c resp;parmchk2 -i $i.prep -f prepi -o $i.frcmod -a y;cd ../;done
复合物体系准备
- 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/ligand/atom_name_check
- # 用师妹的脚本
- # 把 ATOMTYPE.INF ATOMTYPE.INF 复制到各个配体的文件夹下
- # 将d3dock的配体对接结果(sdf文件)转换为pdb文件
- # 2025-3-18 10:30 先用师妹原始的脚本(可处理sdf文件,但是不能自动识别文件夹并处理)
- ls ../g16/>index
- for i in
`cat index`;do mkdir -p $i;cp ../g16/$i/ATOMTYPE.INF $i/;cp ../g16/$i/NEWPDB.PDB $i/;cp ../d3dock/ligprep/${i}.sdf $i/;sed "s/XXXX/${i}.sdf/g" atom_name_check-raw.py >$i/atom_name_check-raw.py;cd $i;python atom_name_check-raw.py;cd ../;done - # 可视化检查没问题
tleap
- # 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/tleap
- # 加载环境
- source ~/zzy/ff19sb.sh
- # 使用我自己编写的脚本
- # 将需要的文件命名为需要的形式即可
- for i in
`cat index`;do mkdir -p $i;cp ../ligand/g16/$i/*frcmod ./$i/lig.frcmod;cp ../ligand/g16/$i/*prep ./$i/lig.prep;cp ../ligand/atom_name_check/$i/LIG.PDB ./$i/lig.pdb;done - (base) [dddc@localhost 91433-17-9-dock-PL]$ cp ../../pro/pdb2pqr/PL_Dimer.pdb ./pro.pdb
- (base) [dddc@localhost 1486-70-0-dock-PL]$ cp ../../pro/pdb2pqr/PL_Dimer.pdb ./pro.pdb
- (base) [dddc@localhost 56083-03-5-dock-PL]$ cp ../../pro/pdb2pqr/PL_Monomer.pdb ./pro.pdb
- (base) [dddc@localhost 56083-03-5-dock-3CL]$ cp ../../pro/pdb2pqr/3CL_Monomer.pdb ./pro.pdb
- (base) [dddc@localhost 59870-65-4-dock-3CL]$ cp ../../pro/pdb2pqr/3CL_Dimer.pdb ./pro.pdb
- for i in
`cat index`;do cp md_parm_gen_chinese.py $i;cd $i;python md_parm_gen_chinese.py 1;cd ../;done - # 蛋白文件手动复制,文件名统一为pro.pdb
- # 可视化检查
- # 56083-03-5两个体系配体坐标信息不对,检查
- # 56083-03-5 二聚体,要检查
amber_to_gmx
- # 23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/sys-pre/amber_to_gmx
- # 使用紫玉的脚本
- # 3CL二聚体两条件需要分别进行位置限制,PL两条链看起来完全一样(system1显示有两个),无需操作
- #################################### do.sh #############################################
- for i in
`cat index` - do
- mkdir -p $i
- cp ../tleap/$i/complex* $i
- cd $i
- cp ../amber_to_gmx-add_restraint.py ./
- python amber_to_gmx-add_restraint.py 0 complex.prmtop complex.inpcrd
- python amber_to_gmx-add_restraint.py 1
- (echo '1|13'; echo 'q') | gmx_mpi make_ndx -f gmx.gro -o index.ndx
- echo 1|gmx_mpi genrestr -f gmx.gro -n index.ndx -o posre1.itp
- cd ../
- done
- ############################################################################################
手动修改:
- 所有的二聚体,都要检查posre1.itp ,其中的原子数与top里 sytem1一样
- 根据配体原子数生成 posre2.itp
- 3CL二聚体有 system1 system2 system3 (二聚体两条链不完全一致)
预平衡
- # 4090 /home/data/zzy/project/hsbd/2025-4bioactive/pre-equ
- # min-nvt-npt.sh 在mdp文件合适的情况下仅需要修改工作路径,就可以自动完成所有体系的预平衡
- dddc@gpu-4090:/home/data/zzy/project/hsbd/2025-4bioactive/pre-equ$ nohup bash min-nvt-npt.sh &
- [1] 2697051
MD模拟任务
- 75.3 /home/dddc/zyzhou/project/hsbd/2025-4bioactive/cmd/100ns/
2025-03-20 5个体系中4个已结束,先进行轨迹分析
91433-17-9-dock-PL 在4090上进行中

轨迹分析
The results are presented in three parts: (i) the “Dynamic Behavior of Complexes” section, which discusses RMSD and RMSF analyses; (ii) the “Predicted Highly Frequent Residues of Ligand Binding” section, which examines the interaction frequencies between ligands and key residues; and (iii) the “Essential Residues in Protein–Ligand Binding” section, which uses MM/GBSA calculations to identify critical binding residues.
周期性处理
- # GMX -pbc nojump -ur compact + -fit rot+trans
- scp dddc@172.21.85.23:/home/databank_70t/zzy/project/hsbd/2024-pl-13mol/analysis/periodic-processing/*sh ./
- # 修改工作路径后,可自动处理轨迹,间隔2000帧输出轨迹可视化结果,需要逐个检查
RMSD
- 75.3 /home/dddc/zyzhou/project/hsbd/2025-4bioactive/cmd/analysis/rmsd
- scp dddc@172.21.85.23:/home/databank_70t/zzy/project/hsbd/2024-pl-13mol/analysis/rmsd/complex/* ./
- # 之前有写过脚本,可直接使用,需要修改路径
- # 作图脚本需要逐行检查
- # 已出图,没问题
RMSF
- 75.3 /home/dddc/zyzhou/project/hsbd/2025-4bioactive/cmd/analysis/rmsf
- cp ../rmsd/* ./
- # 与rmsd的脚本逻辑几乎完全一致,rmsf的gmx计算命令稍有不同,修改即可
- # 作图脚本需要逐行检查
- # RMSF是根据残基计算的(-res),在处理双链时需要注意,两条链需要分开处理
- # 本身也是很有意思的信息,双链RMSF波动的区别可以一定程度上反应配体的结合吗?
- # 看了一下,两条链中差距不算大
- # 分别做了两条链按照残基取平均的结果和比较每次平行实验两条链RMSF的图
- # 两条链取平均后,三次实验再取平均的图(简单说明体系稳定)还没做
MM/GBSA
- 85.23 /home/databank_70t/zzy/project/hsbd/2025-4bioactive/analysis/mmgbsa
- (base) [dddc@localhost workdir]$ nohup bash mmgbsa-cal.sh &
- [1] 213572
- # 已在23上提交任务 18:28
- # 尝试在75.3上安装amber24
- # /home/dddc/zyzhou/software/amber24/amber24_src
- # ./update_amber --update 已完成 18:32