
D3MD
- # 测试路径 172.21.75.1 /home/dddc/zzy/test/0515-gmx/test-grompp
- # 按时间合并轨迹的方法:
- # 在提交每一次独立的短时MD任务时,在生成tpr文件时,在mdp文件中加一行“tinit”参数(单位 ps),指定模拟开始时间
- # gro文件修改(若符合交换条件,交换速度),作为下一次模拟的参考构象(-c);下一次模拟需要新的mdp文件,mdp 文件中 添加“tinit”参数 单位 ps,指定开始时间,生成tpr
- # 最后,用gmx的trjcat命令合并所有的轨迹(-f 后可以接任意数量的轨迹)
- gmx_mpi trjcat -f 1.xtc 2.xtc -o merge.xtc
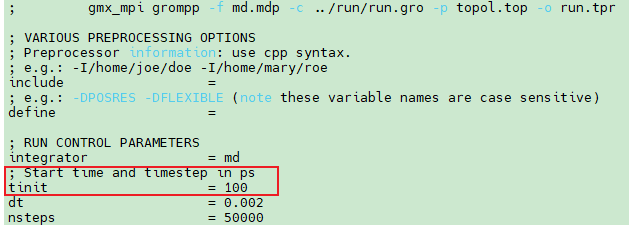
用户需要准备 与 副本数相对应的mdp文件,每个mdp文件中仅是温度与”tinit”参数不同
在GPU上跑gromacs, mdp 文件中 能量组改成system
1. 调用md生成tpr文件
- # -maxwarn 1 感觉可以不加,加了就是增加容错。正常情况下尽可能调整至无warning。
- gmx_mpi grompp -f md.mdp -c start.gro -p topol.top -n index.ndx -o run.tpr -maxwarn 1
2. gromacs GPU cMD 2ps
- # 这一步 deffnm 后的名字得和上面tpr 的名字保持一致,跑出来的文件名字(run.gro ; run.xtc ; run.log 等)也都是这个
- gmx_mpi mdrun -v -deffnm run
3. gmx CPU 计算能量
- # 基于上一步动力学得到的最后一帧gro。提取出需要计算的几个对象(蛋白,小分子,溶剂)
- # echo 后面跟的数值是 选择对应的组, 不同体系之间可能略有差异
- echo 1|gmx_mpi trjconv -s run.tpr -f run.gro -o pro.pdb
- echo 13|gmx_mpi trjconv -s run.tpr -f run.gro -o mol.pdb
- echo 17|gmx_mpi trjconv -s run.tpr -f run.gro -o sol.pdb
- # 若需要 自定义 组 (例如: 蛋白-小分子)可生成对应的index文件,见“自定义组(index)”部分
- # 分别生成他们各自的tpr
- # echo 后面跟的数值是 选择对应的组, 不同体系之间可能略有差异
- echo 1|gmx_mpi convert-tpr -s run.tpr -o pro.tpr
- echo 13|gmx_mpi convert-tpr -s run.tpr -o mol.tpr
- echo 17|gmx_mpi convert-tpr -s run.tpr -o sol.tpr
- # 分别计算能量, 这一步输出的都叫 md.log ,得改名字,不然会覆盖
- gmx_mpi mdrun -s pro.tpr -rerun pro.pdb
- mv md.log pro.log
- gmx_mpi mdrun -s mol.tpr -rerun mol.pdb
- mv md.log mol.log
- gmx_mpi mdrun -s sol.tpr -rerun sol.pdb
- mv md.log sol.log
- # 在输出的log文件里提取能量



自定义组(index)
- gmx_mpi make_ndx -f gromacs.gro -o index.ndx
- # 若需要选取多个 用 | 连接即可,例如下图
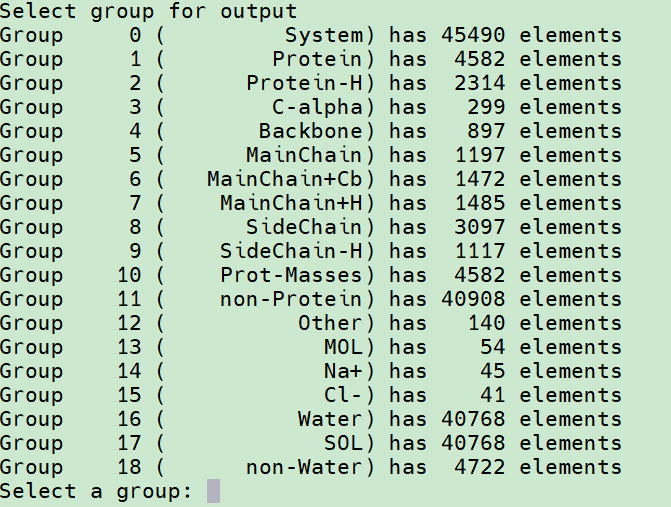

合并轨迹
merge_traj.sh
- cpptraj <<EOF
- parm ref.pdb
- trajin 1.xtc
- trajin 2.xtc
- trajin 3.xtc
- autoimage
- rms
- trajout ${i}_autoimage_fit.xtc
- trajout ${i}_look.pdb offset 1000
- run
- EOF
- done