
smina-盲对接
思路: 把整个蛋白质装进对接盒子,允许在整个盒子中搜索小分子构象
对接中心确认:
pymol中切换-chains;选中整个蛋白质;命令行计算质心:
- centerofmass sele
输出案例如下图:
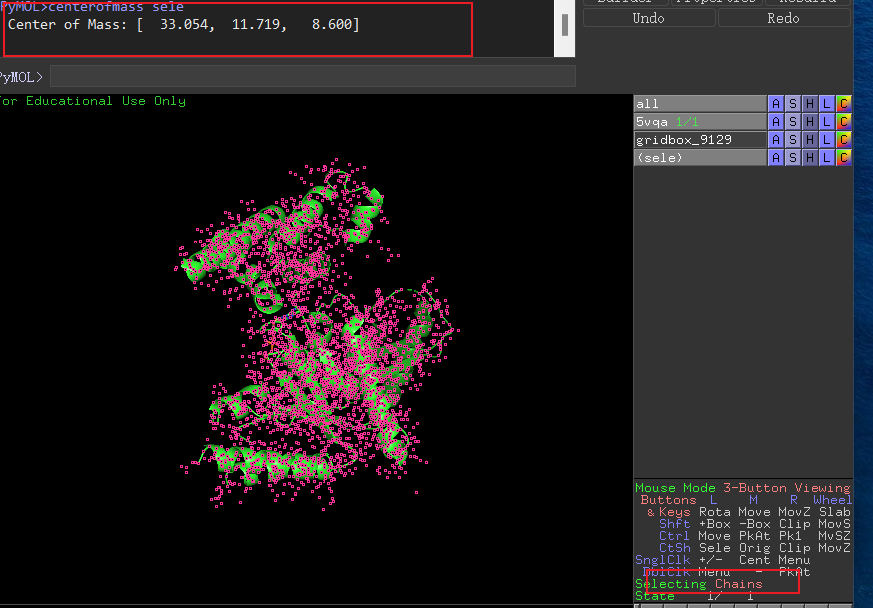
得到的三个值分别为 smina对接命令里的 --center_x / --center_y / --center_z 后跟的值
对接盒子尺寸确认
有现成的脚本:原链接: DrawGridBox - PyMOLWiki
下载后将脚本“drawgridbox.py” 拖入pymol
- load drawgridbox.py
- # 命令的参数可见链接DrawGridBox - PyMOLWiki;一般改一下 ${your_protein_name}就行,其他参数默认
- drawgridbox ${your_protein_name}, nx=5, ny=5, nz=5, lw=1, g=0, b=0
- drawgridbox ${your_protein_name}, nx=5, ny=5, nz=5, padding=5, lw=1, r=0, g=0
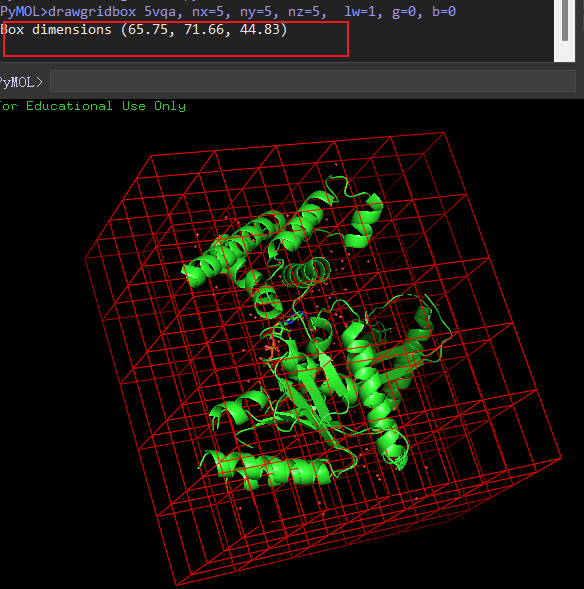
输出案例如下图所示:
脚本会自动生成这个红色的盒子,对smina对接而言,有用的是红框里的三个数值,代表盒子尺寸
即 smina对接命令里的 --size_x / --size_y / --size_z 后跟的值
对接
与常规smina对接无异;只是 --center_x / --size_x 20 (y;z) 后面的值换成前两步得到的值
- # 对接命令案例 smina -h 可查看参数
- smina --seed 1 --num_modes 10 -r 6lk0fix-md.pdb -l 0415.pdb --center_x 48.997 --center_y 44.261 --center_z 37.907 --size_x 20 --size_y 20 --size_z 20 -o dock_0415-t13.pdb