网药-案例share
2024-12-09
成分信息收集
根据化湿败毒方中的14味中药材,收集2049个成分
- hsbd-2049mol.smi
靶点信息收集
- # ETCM数据库 寻找他们的已知靶标(确证+基于配体相似性的潜在靶点)
- # 85.3 /home/yqyang/zzy/hsbd/np/mgp/mol-targets/hsbd-mols-targets
- # do2.sh 的逻辑如下:
- 1. 根据 hsbd-2049mol.smi 中的smi,去数据库(etcm-mols-smi-targets-fyl.csv)中匹配smi,若得到了唯一的匹配结果,则进行后续操作(929/2049);
- 2. 判断数据库中,该靶点是否具有确证靶点,若有,将靶点信息记录至mol-targets-name/$a.txt(文本处理,使得每一行仅有一个靶点名称),进一步的,根据etcm-targets-yls.csv,将靶点名称替换为UniProt ID(需要名称-ID之间是唯一对应),结果记录至 mol-targets-id/$a.txt;
- 3. 判断数据库中,该靶点是否具有潜在靶点(基于配体相似性的预测方法),其余步骤与步骤二一致。
- # 85.3 /home/yqyang/zzy/hsbd/np/get-hsbd-mols-targets/all-2049
- HSBD consists of 14 herbs, and we can find corresponding information for 2049 compounds in the ETCM2.0 database. Furthermore, we found that 929 of the compounds match perfectly (the grep result returns only one line), with 510 hsbd-targets.txt corresponding targets.
- # 85.3 /home/yqyang/zzy/hsbd/np/get-hsbd-mols-targets/insert
- # 从Genecards上下载“SARS-CoV-2”相关蛋白(excel),共7096个有UniProt ID
- # 与HSBD-D3AI 成分潜在靶点取交集
- cat genecards-SARS-CoV-2.txt hsbd-targets.txt |sort|uniq -d >insert
- (base) [yqyang@localhost insert]$ wc -l *
- 7095 genecards-SARS-CoV-2.txt
- 510 hsbd-targets.txt
- 236 insert
- 7841 总用量
PPI
- # insert对应的236个UniProt ID 直接输出strings (选择人源)
成分-靶点-通路网络图
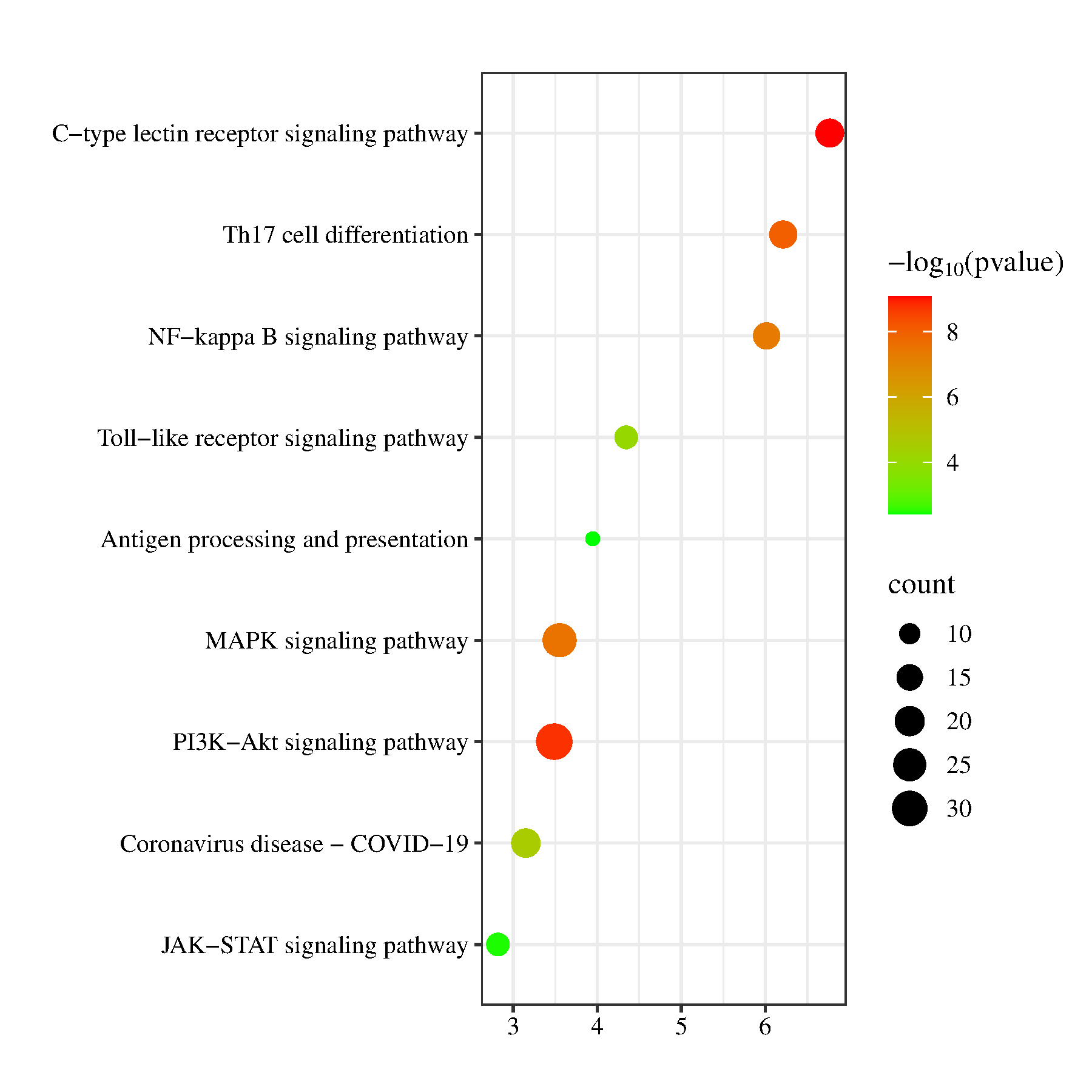
根据PPI网络图中展示的靶点,去找对应的成分,再找对应的通路,参考连钱草课题的流程
236个靶点输入 KEGG DAVID:DAVID: Functional Annotation Result Summary (ncifcrf.gov)。KEGG pathway富集分析得到了167个通路,让Ghatgpt帮我选出了9个与新冠治疗相关的通路。
将KEGG pathway富集分析的原始结果文件整理为以下格式:
第一列:序号;
第二列:通路名称;
第三列:通路对应的靶点。
上传服务器,运行脚本 mol-gene-pathway.sh
- # 85.3 /home/yqyang/zzy/hsbd/np/mgp/mol-gene-pathway.sh
- # 该脚本的逻辑如下:
- 1. 根据整理后的KEGG结果文件,构建 通路-靶点关联;
- 2. 根据 去重后的靶点信息,找到能作用于这些靶点的成分;
- 3. 大量的文本操作,输出固定格式的文件。
- # 化合物数量过多,做了一些限制
- 1. 修改输入数据:舍去潜在靶点数小于10的化合物,还剩218个
- for i in
`ls mol-targets-id`; do - line_count=$(wc -l < mol-targets-id/$i) # 获取文件行数
- echo "$i has $line_count lines" # 打印文件名和行数
- # 如果文件行数小于 10,则删除该文件
- if [ "$line_count" -lt 10 ]; then
- echo "Deleting file: mol-targets-id/$i"
- rm mol-targets-id/$i
- fi
- done
- 2. 修改脚本mol-gene-pathway.sh:舍去对应的化合物数量小于10的靶点(这部分靶点不查找对应化合物)
- for a in
`awk '{print $2}' ${pathway_num}pathway-${gene_num}gene.txt|sort|uniq` - do
- #grep -w "$a" mol-targets-id/TCMID*|sed 's/mol-targets-id\///g'|sed 's/.txt:/ /g'>>test
- # 查找每个分子对应的基因
- genes=$(grep -w "$a" mol-targets-id/TCMID* | sed 's/mol-targets-id\///g' | sed 's/.txt:/ /g')
- # 统计该分子对应的基因数量
- gene_count=$(echo "$genes" | wc -l)
- # 如果基因数量 >= 10,保存到 test 文件
- if [ "$gene_count" -ge 10 ]; then
- echo "$genes" >> test
- fi
- done
- 3. 实际操作中,上述两个方法效果都没有有效减少成分数量,故采用以下步骤
- # 统计原始输出文件9pathway-78gene-184mol.txt中每个成分的degree
- for i in
`grep mol label.txt |awk -F ',' '{print $1}'`;do num=`grep -w ${i} ../9pathway-78gene-184mol.txt |wc -l`;echo "$i $num">>111;done - # 大概看一下degree 分布
- sort -k2,2nr 111
- # 选出degree小于4的化合物
- awk '$2<4{print $1}' 111 >degree_less_than_4_mol
- # 把这些化合物从 9pathway-78gene-184mol.txt删除,得到9pathway-78gene-32mol.txt
- for i in
`cat degree_less_than_4_mol`;do sed -i "/$i/d" 9pathway-78gene-184mol.txt;done - mv 9pathway-78gene-184mol.txt 9pathway-78gene-32mol.txt
得到的9pathway-78gene-32mol.txt与label.txt输入cys软件
输入的时候,要指定 source node 和 target node 且 指定分隔符
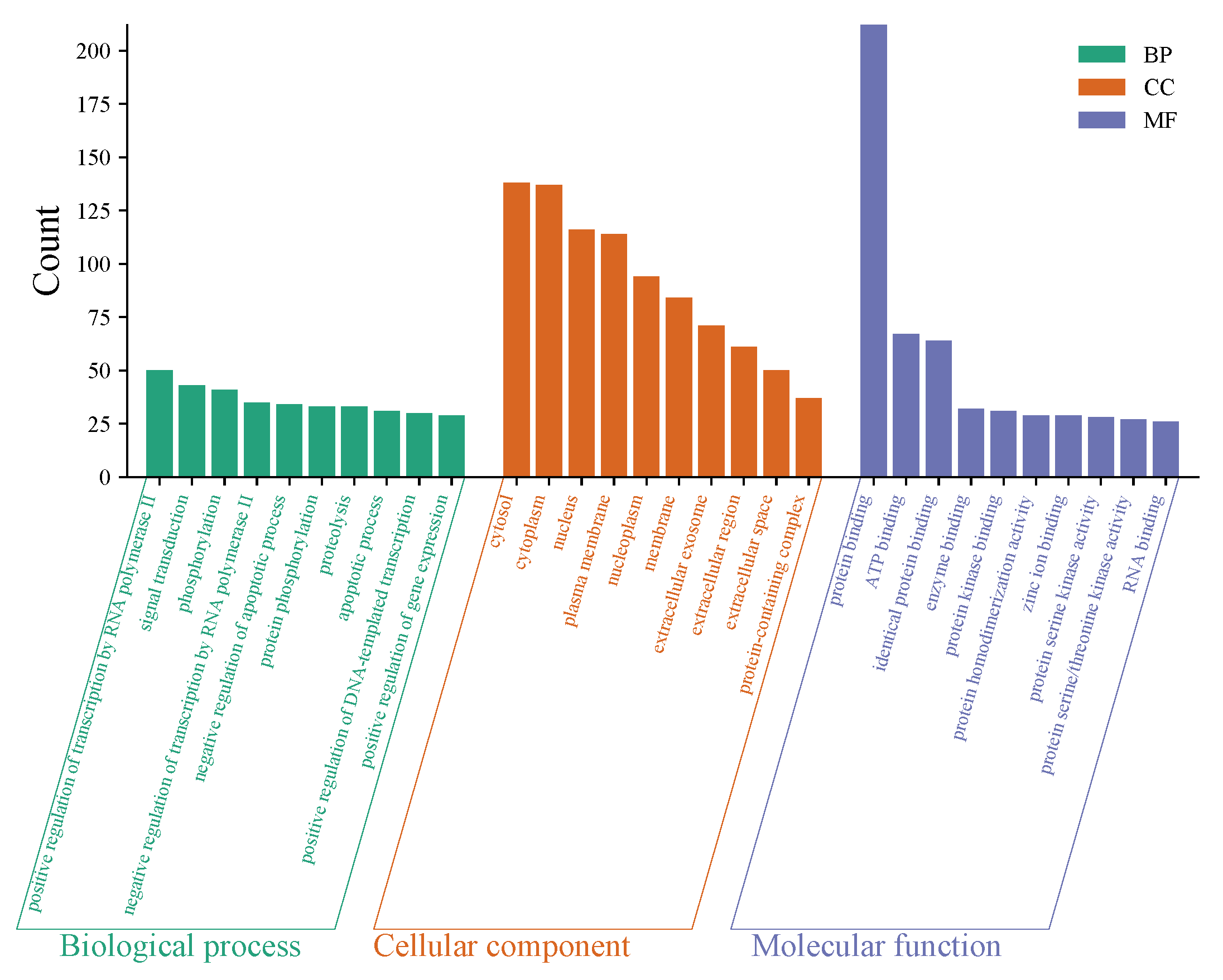
GO/KEGG分析
david数据库DAVID: Functional Annotation Result Summary (ncifcrf.gov) 中获取数据。
上传 insert,选择Uniprot_accession,提交任务。
选择gene_ontology下载go分析需要的文件,BP,CC,MF都要。
链接另存为txt文件,导入excel , 选有效数据 (Term; RDR; Pvalue; Count) 另存为文本文件。
导入微生信平台,出图。
微生信平台有示例数据,在此基础上将GO通路,y轴(第三列)数据替换为自己的即可。