
vsREMD 操作
确定需要的vsREMD副本数



/home/databank/zzy/usp18/vsREMD/noloop_24/parameters
初始结构准备
注1:若是每个副本构象不同,可参考该流程,若初始结构完全一致,可跳转至“vsREMD测试”部分
注2:每个副本构象不同理论上更有利于充分采样
- ### tleap
- # 把tleap的操作流程写入tleap${i}.in,记得更改每个tleap.in里的蛋白名称
- # tleap${i}.in 大致模板如下
- ###########################################################################################
- source leaprc.protein.ff19SB
- source leaprc.water.opc
- pro=loadpdb pM.pdb
- charge pro
- solvatebox pro OPCBOX 10
- addionsrand pro Na+ 4
- charge pro
- saveamberparm pro pro.prmtop pro.inpcrd
- savepdb pro pro_tleap.pdb
- ###########################################################################################
- # 记得到不同的目录下执行
- tleap -s -f tleap${i}.in
- python change.py
- ####### change.py ###################################################
- import parmed as pmd
- amber = pmd.load_file('pro.prmtop','pro.inpcrd')
- amber.save('topol.top')
- amber.save('gromacs.gro')
- #####################################################################
- # 添加位置限制信息
- !!!! vsREMD的 topol文件;posre.itp;index.ndx 不同副本需要一致,这意味着原子数需要完全一致,但是tleap加溶剂往往数目不会完全一致,因此需要选择一个副本作为基准(一般是溶剂数最少的),所有副本都用它的topol文件;posre.itp;index.ndx。手动删除其他副本gromacs.gro里的多余溶剂分子,使其原子数一致。
- # 案例:一个有20个副本的体系,以副本6为基准,原子数59872,溶剂数14035
- # 手动删除其他副本的溶剂至14035后,还要更改gromacs.gro第二行的总原子数
- for a in {1..25};do sed -i "2s/.*/59872/" ../${a}/gromacs.gro;done
- # 用副本6的gromacs.gro的文件生成
- # 事实上,用哪一个副本都是一样的
- gmx_mpi make_ndx -f gromacs.gro -o index.ndx
- gmx_mpi genrestr -f gromacs.gro -o posre.itp
预平衡(for loop)
- # 300K 平衡
- # 模板如下
- # 记得检查gmx版本
- which gmx_mpi
- ###########################################################################################
- for i in {1..20}
- do
- cd ${i}
- gmx_mpi grompp -f ../parameters/minim.mdp -c gromacs.gro -p ../topol_6/topol.top -o em.tpr
- mpirun -np 50 gmx_mpi mdrun -v -deffnm em
- gmx_mpi grompp -f ../parameters/nvt.mdp -c em.gro -r em.gro -p ../topol_6/topol.top -o nvt.tpr
- mpirun -np 50 gmx_mpi mdrun -v -deffnm nvt
- gmx_mpi grompp -f ../parameters/npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p ../topol_6/topol.top -n ../topol_6/index.ndx -o npt.tpr
- mpirun -np 50 gmx_mpi mdrun -v -deffnm npt
- done
- ###########################################################################################
- # 记得检查gmx版本
- which gmx_mpi
- # 升温平衡
- # 将mdp文件中的温度改成vsREMD对应的温度
- for a in {1..20};do b=
`sed -n "${a}p" ../parameter/temp`; sed "s/300/${b}/g" ../parameter/npt.mdp > npt_mdp/npt${a}.mdp; done - # 记得检查gmx版本
- which gmx_mpi
- for i in {1..20};do gmx_mpi grompp -f npt_mdp/npt${i}.mdp -c ../pre/pre${i}.gro -r ../pre/pre${i}.gro -p ../parameter/topol.top -n ../parameter/index.ndx -o npt${i}.tpr -maxwarn 1; done
- # 可以交后台
- nohup sh -c 'for i in {0..25};do mpirun -np 50 gmx_mpi mdrun -v -deffnm npt${i}; done' &
vsREMD测试
- # 1ns test
- for a in {1..20};do b=
`sed -n "${a}p" ../parameter/temp`; sed "s/300.00/${b}/g" ../parameter/md.mdp > md_mdp/md${a}.mdp; done - for i in {1..20};do gmx_mpi grompp -f md_mdp/md${i}.mdp -c ../npt/npt${i}.gro -p ../parameter/topol.top -n ../parameter/index.ndx -o md${i}.tpr -maxwarn 1; done
- nohup mpirun -np 100 gmx_mpi mdrun -v -deffnm md -multidir md1 md2 md3 md4 md5 md6 md7 md8 md9 md10 md11 md12 md13 md14 md15 md16 md17 md18 md19 md20 -replex 1000 &
- #查看任务
- 1.tail -f nohup.out
- 2.jobs
- 3.vi nohup.out
- 4.vi md0.log
调整温度参数
对于交换率不满足0.2-0.5的副本,需要调整温度间隔
若<0.2,调小T间隔
若大于0.5,调大T间隔
测试结果实例:

自动温度调参脚本路径
- 24 /home/databank/zzy/people/xcy/vsremd-20250905/test/vsREMD_temp_adjustment.sh
脚本使用方法简述
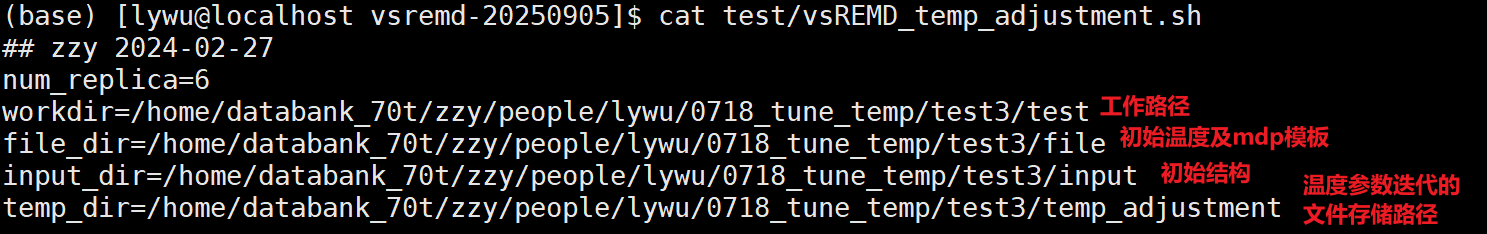
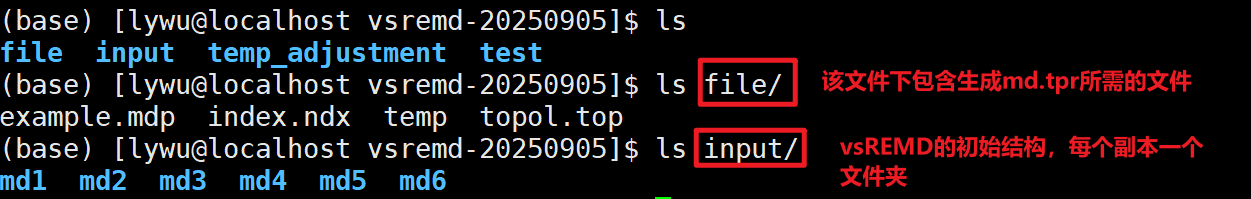
input 文件夹里就放初始温度列表下,vsREMD测试跑完的结果即可
