
9.25 SMD test
9.24 的测试中发现了一些问题:
首先,之前的SMD指定了牵引的维度(z轴),这样可以将配体拉的更远一些,因为牵引距离不得超过盒子大小的一半的这个距离是按照这个来定义的,三个维度都放开肯定比一个维度值更大,换言之,更快的到达牵引距离限度。但是可视化检查了一下,欧几里得距离达到盒子大小的一半足够了(30nm左右),根据师姐目前PMF测试的结果,低能点在10-15A,高能点在20-25附近,足够了。
经测试,这样 smd_pullx 可以直接拿来作图了
k/rate test
- # force vs. Time
- 1. 每个参数平行三次的结果统计
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1000
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1250
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1500
- # 最后结束的时间实际上是不一样的,这里为了统一,只统计到最短的那个结束时间
- paste */smd_pullf.xvg|sed '1,17d'|sed '1i time1 f1 time2 f2 time3 f3'|awk '$6 != ""' >smd_pullf.xvg
- 2. 不同参数合起来统计
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005
- paste */smd_pullf.xvg|sed '1d'|awk '{print $1,$2,$4,$6,$8,$10,$12,$14,$16,$18}'|sed '1i time f1 f2 f3 f1 f2 f3 f1 f2 f3'|awk '$10 != ""' >smd_pullf.xvg
- # dis vs. Time
- 1. 每个参数平行三次的结果统计
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1000
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1250
- home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005/1500
- # 最后结束的时间实际上是不一样的,这里为了统一,只统计到最短的那个结束时间
- paste */smd_pullx.xvg|sed '1,17d'|sed '1i time1 dis1 time2 dis2 time3 dis3'|awk '$6 != ""' >smd_pullx.xvg
- 2. 不同参数合起来统计
- /home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0925/k_rate_0.005
- paste */smd_pullx.xvg|sed '1d'|awk '{print $1,$2,$4,$6,$8,$10,$12,$14,$16,$18}'|sed '1i time dis1 dis2 dis3 dis1 dis2 dis3 dis1 dis2 dis3'|awk '$10 != ""' >smd_pullx.xvg
较方便的流程化跑测试的脚本
/home/dddc/zzy/project/koff/smd/param-test/trypsin-31/0925/k_rate_0.005/run.sh
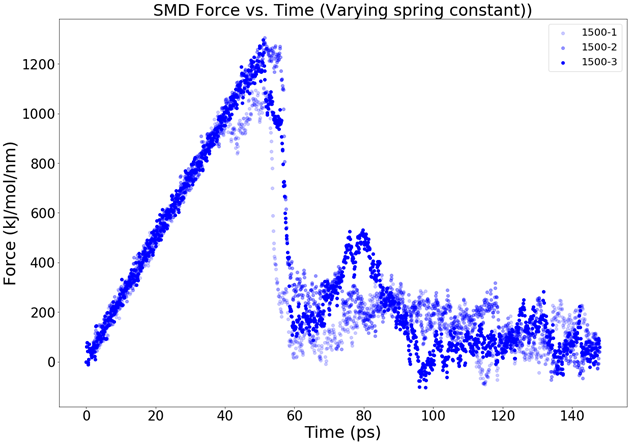
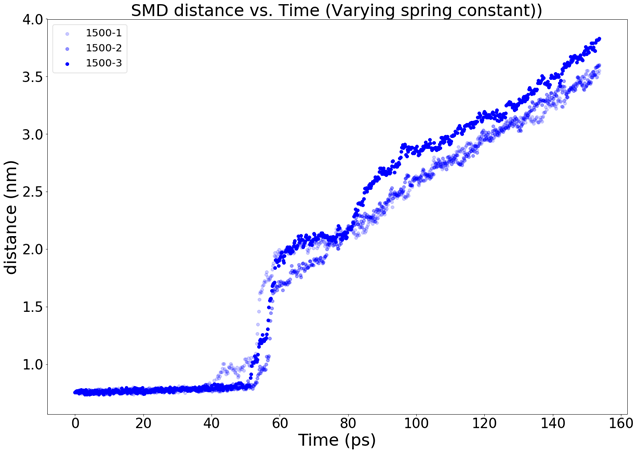
现在能较好的进行测试,作图了,能比较好的比较 不同 k rate 下的 force/cv - time 曲线
但是问题是,我们的目的是从SMD中挑选vsREMD的初始构象,换言之,得测试才能知道这个初始构象好不好
从 force/cv - time 曲线 里,我们都只能推断 这些构象大概长啥样 ,结合可视化可以进一步”喔~长这样“,但是 能不能算出好的PMF呢,不知道。
综上所述,先把重心放到 后面,从SMD中 流程化,可重复的 挑选vsREMD的初始构象上
SMD-vsREMD inital confs
- # 测试所用SMD 轨迹
- /home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0924/k/1500/3
跟Leyun 讨论后,初步想法是,从fmax往后,一直到 蛋白-配体 mindist 大于等于4A
这段轨迹提取出来,聚类。规定聚类个数等于副本数。每个类等于
轨迹提取
- # 定位 fmax (单位为ps)
- sort -g -k2nr ../smd_0.02_1500/smd_pullf.xvg|head -n5
- # 定位 配体解离 (蛋白-配体 mindist 大于等于4A)
- echo 1 13|gmx_mpi mindist -s ../smd_0.02_1500/dry.pdb -f ../smd_0.02_1500/dry.xtc
- awk '{printf "%f %f\n", $1, $2}' mindist.xvg|awk '$2>0.4{print}'|head -n5
- # 提取轨迹
- gmx_mpi trjconv -s ../smd_0.02_1500/dry.pdb -f ../smd_0.02_1500/dry.xtc -b 51.0000 -e 149.600000 -o dis-process.xtc
- gmx_mpi trjconv -s ../smd_0.02_1500/smd.tpr -f ../smd_0.02_1500/smd.xtc -b 51.0000 -e 149.600000 -o dis-process.xtc
- gmx_mpi trjconv -s ../smd_0.02_1500/smd.tpr -f ../smd_0.02_1500/smd.xtc -b 51.0000 -e 149.600000 -o ref.pdb -dump 0
- # 老规矩,可视化检查
- gmx_mpi trjconv -s ../smd_0.02_1500/dry.pdb -f ../smd_0.02_1500/dry.xtc -b 51.0000 -e 149.600000 -o look.pdb
- # 流程化
- fmax-ps=
`sort -g -k2nr ../smd_0.02_1500/smd_pullf.xvg|head -n1|awk '{print $1}'` - echo 1 13|gmx_mpi mindist -s ../smd_0.02_1500/dry.pdb -f ../smd_0.02_1500/dry.xtc
- dis-ps=
`awk '{printf "%f %f\n", $1, $2}' mindist.xvg|awk '$2>0.4{print $1}'|head -n1`
聚类
- 85.24 /home/databank/zzy/project/MD/koff/smd/vsREMD_inital_confs/trypsin-benzamidine/test1/cluster/
- # 因为要指定聚成 X 类
- cpptraj
- > parm /home/databank/zzy/project/MD/koff/smd/vsREMD_inital_confs/trypsin-benzamidine/smd_0.02_1500/dry.pdb
- > trajin /home/databank/zzy/project/MD/koff/smd/vsREMD_inital_confs/trypsin-benzamidine/test1/dis-process.xtc
- > cluster C1 kmeans clusters 20 randompoint maxit 500 repout sieve 10 random rep repfmt pdb summary summary.dat
- > run
- # test1
- # 指定
- # test1 是kmeans 聚类,test2换成了 hieragglo,20个构象差异不大,分布更合理了
- # 但是,这两次测试都发现,不写distance metric那一行才能得到合理的结果
- # cpptraj parminfo 检查发现,一共224个氨基酸,换言之,配体不能以氨基酸的形式表示,遂更换为 原子,可以进行聚类
- ###################################################################################
- cpptraj <<EOF
- parm $dir/dry.pdb
- trajin $dir/dis-process.xtc
- cluster C1 \
- hieragglo epsilon 3.0 clusters 20 averagelinkage \
- rms @3221-3238 \
- sieve 10 random \
- summary summary.dat \
- repout rep repfmt pdb
- run
- EOF
- ##################################################################################
- # 结果很合理了!再试试能不能将distance metric换成 反应坐标(三个蛋白质中原子 2465,2467,2475 质心和配体质心距离)
- # 经过漫长的尝试,拿到了结果,还是得看官方教程,害
- # git上的cpptraj,有一个最新的doc,里面很全面,给出了如何
- ###################################################################################
- cpptraj <<EOF
- parm $dir/dry.pdb
- trajin $dir/dis-process.xtc
- distance key_resn_bb_atom_to_MOL @2465,2467,2475 @3221-3238
- cluster \
- hieragglo epsilon 3.0 clusters 20 averagelinkage \
- data key_resn_bb_atom_to_MOL \
- sieve 10 random \
- summary summary.dat \
- repout rep repfmt pdb \
- avgout avg avgfmt pdb
- run
- EOF
- ##################################################################################
- # 记得用原始轨迹(处理周期性,保留整个体系,不要dry)
打包
- trypsin-benzamidine 20240927新一轮vsREMD初始构象(晶体结构-SMD-聚类)
- 85.24 /home/databank/zzy/project/MD/koff/smd/vsREMD_inital_confs/trypsin-benzamidine/cluster (参数文件 在params 文件夹下)
- # 本轮初始构象参数如下:
- 1. SMD
- 4090 /home/dddc/zzy/project/koff/smd/param-test/trypsin-benzamidine/0924/k/1500/smd.mdp
- 1)初始构象:晶体结构;
- 2)反应坐标:ASP171 backbone 原子(a_2465_a_2467_a_2475);
- 3)位置限制:蛋白backbone
- 4)弹簧牵引速率:0.02 nm per ps;弹簧力常数:1500 kJ mol^-1 nm^-2
- 2. 聚类
- 85.24 /home/databank/zzy/project/MD/koff/smd/vsREMD_inital_confs/trypsin-benzamidine/cluster
- 1)Distance Metric Options: distance key_resn_bb_atom_to_MOL @2465,2467,2475 @3221-3238
- 2)Algorithms: hieragglo epsilon 3.0 clusters 20 averagelinkage
- 3)输入轨迹:SMD fmax-反应坐标(距离)超过4A的第一帧(-b 51.0000 -e 149.600000)
- # test 文件夹下测试了一下tpr打包,没问题
- gmx_mpi grompp -f ../params/md.mdp -p ../params/gmx.top -n ../params/index.ndx -r ../rep.c0.pdb -c ../rep.c0.pdb -o md.tpr