
8.19 batch3
体系挑选
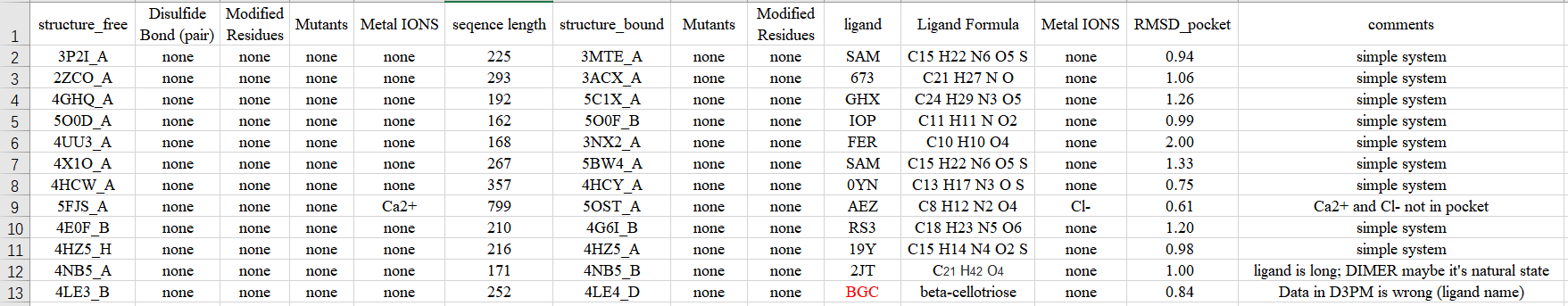
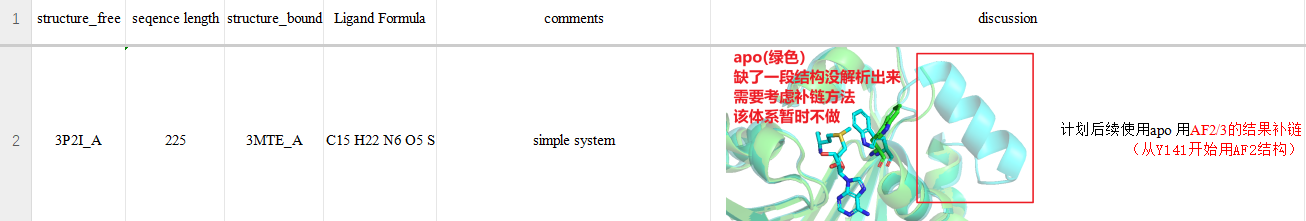
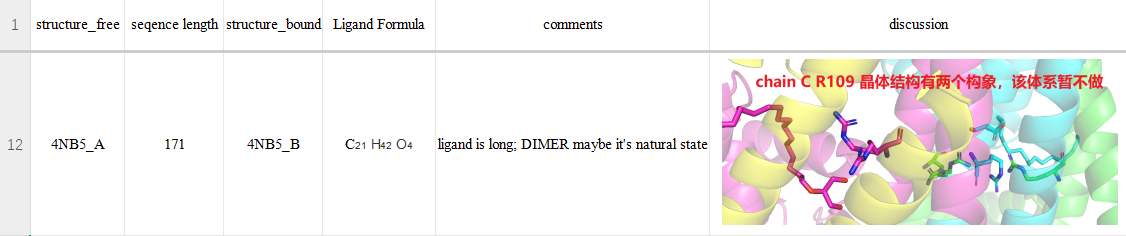
讨论
有三个体系暂且不做

蛋白检查 & 清洗
PDBfixer
- 75.5 /home/databank/zyzhou/project/d3pm/batch3/protein_preparation
- # 将需要下载的pdb id 上传pdb网站,进行批量下载,解压后
- # 文件放于/home/databank/zyzhou/project/d3pm/batch3/protein_preparation/pdb
- # /home/databank/zyzhou/project/d3pm/batch3/protein_preparation/pdbfixer
- # Pdbfix 只要给定了--pdbid 参数,可以自己从pdb网站获取输入结构
- # --add-residues 可以将氨基酸序列补齐,但是也会将两端补上,这往往会导致错误
- for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do pdbfixer --pdbid=${i} --ph=7.0 --keep-heterogens=none --add-atoms=all --add-residues --output=${i}-pdbfixer.pdb;done - # 将不补链的结果存储在no_add-residues文件夹
- # 将补链的结果存储在add-residues文件夹
统一氨基酸序列
- # 撰写脚本 do.sh,统计两端补链前后的变化,N端的补链均已删除(已晶体结构为准)
- # C端的未作删除,变化情况记录在nohup.out中
- # 检查N端,apo-complex 需要保证至少氨基酸序列一致
- for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do echo "########################## $i ##########################";head -n1 ${i}*;done - # 手动将apo-complex的C,N端开始,结束原子统一,规则如下:
- 1)以 apo/complex 中N端更小序列为准进行保留;
- 2)以 apo/complex 中C端更大序列为准进行保留; 定位到需要保留的最后一个氨基酸。分情况讨论:
- 情况1:该氨基酸即为不进行缺失氨基酸补齐状态下(no_add-residues文件夹)的最后一个氨基酸:
- 用不进行缺失氨基酸补齐状态下的最后一个氨基酸坐标(已封端)替换
- 情况2:该氨基酸不是不进行缺失氨基酸补齐状态下(no_add-residues文件夹)的最后一个氨基酸:
- 仅需删除多余的氨基酸,不进行封端(会少一个OXT原子,让tleap去补)
pdb2pqr加氢
- # 75.5 /home/databank/zyzhou/project/d3pm/batch3/protein_preparation/pdb2pqr
- ############################## do.sh #########################################################
- for i in {1..18}
- do
- a=
`sed -n "${i}p" ../list|awk -F '_' '{print $1}'|tr '[:upper:]' '[:lower:]'` - b=
`sed -n "${i}p" ../list|awk -F '_' '{print $2}'` - echo "########################## ${a} chain ${b} ##########################"
- awk '$12!="H"{print}' ../pdbfixer/${a}_${b}-pdbfixer-clean.pdb >${a}_${b}-noH.pdb
- pdb2pqr30 ${a}_${b}-noH.pdb ${a}_${b}.pqr --ff AMBER --ffout AMBER --with-ph 7.0 --pdb-output ${a}_${b}-pdb2pqr.pdb
- rm ${a}_${b}-noH.pdb ${a}_${b}.pqr
- done
- ###############################################################################################
配体处理
- 23 /home/databank_70t/zzy/g16/D3PM/batch3
- # 9个体系,直接去pdb页面上下载sdf文件
- 23 /home/databank_70t/zzy/g16/D3PM/batch3/epik
- # 2021 薛定谔ligprep epik 对其进行质子化
- bash do.sh
- 23 /home/databank_70t/zzy/g16/D3PM/batch3/
- for i in {2..18..2}; do a=
`sed -n "${i}p" system-name|awk '{print $2}'`;b=`sed -n "${i}p" system-name|awk '{print $1}'`;mkdir -p system${i};cp epik/${a}-lig.sdf system${i}/;done - # 在 75.2 上提交任务
- 75.2 /home/databank/zyzhou/project/D3PM/g16/batch3/g16_cal
- for i in {2..18..2}; do a=
`sed -n "${i}p" ../system-name|awk '{print $2}'`;b=`sed -n "${i}p" ../system-name|awk '{print $1}'`;mkdir -p system${i};cp ../epik/${a}-lig.sdf system${i}/;done - # 检查是否有报错体系
- python check.py -i /home/databank/zyzhou/project/D3PM/g16/batch3/g16_cal -t sdf
- # 均通过
- date | sed "s/^/D3PM system: nine g16 jobs submitted on /" >>nohup.out
- nohup python lig_parameter_cal.py -i /home/databank/zyzhou/project/D3PM/g16/batch3/g16_cal -t sdf
整理装包
- 75.5 /home/databank/zyzhou/project/d3pm/batch3/protein_preparation/clean_system
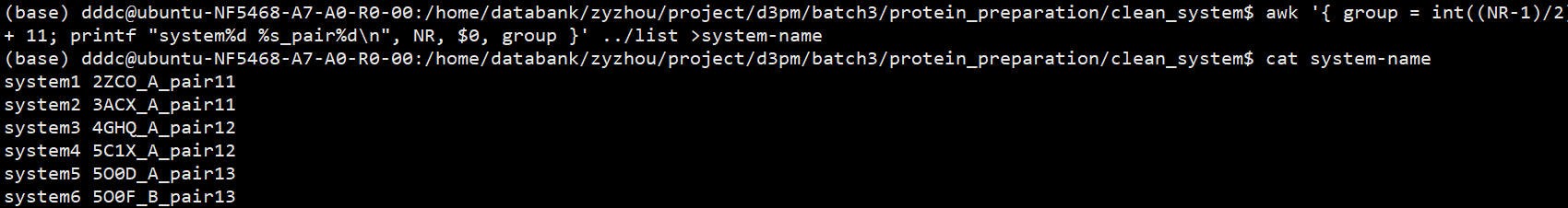
apo体系
- # 炫酷命令,分配好system名字和pair名字
- # batch1,batch2 各搞了5个pair,虽然有些体系在后续的操作中被废弃了,先这么命名
- # system 每次都重新命名
- awk '{ group = int((NR-1)/2) + 11; printf "system%d %s_pair%d\n", NR, $0, group }' ../list >system-name
- for i in {1..18};do a=
`sed -n "${i}p" system-name|awk '{print $2}'|awk -F '_' '{print $1}'|tr '[:upper:]' '[:lower:]'`;b=`sed -n "${i}p" system-name|awk '{print $1}'`;c=`sed -n "${i}p" system-name|awk '{print $2}'|awk -F '_' '{print $2}'`;echo "########## $a $b ###########";mkdir -p $b;cp ../pdb2pqr/${a}_${c}-pdb2pqr.pdb $b/;done
complex体系
- # 75.2 /home/databank/zyzhou/project/D3PM/g16
- # 从75.2将高斯优化结果复制过来
- rsync -rzP batch3 172.21.75.5:/home/databank/zyzhou/project/d3pm/batch3/protein_preparation/clean_system/
- # 75.5 将该文件夹重命名为 g16_from_75.2
- # 75.5 /home/databank/zyzhou/project/d3pm/batch3/protein_preparation/clean_system
- for i in {2..18..2};do cp -r g16_from_75.2/g16_cal/system${i}/* system${i};done
- # 将clean_system文件夹同步至v100进行MD 自动化处理

v100 环境重配
- # 2025-8-24 由于v100的conda转为miniforge,导致很多包要重新安装
- # 将脚本一些基础包安装至 miniforge 环境 zzy
- mamba create -n zzy python=3.8 numpy pandas matplotlib
- # gmx 依赖包要手动加载
- export LD_LIBRARY_PATH=/home/chpeng/software/miniforge3/envs/gcc_12/lib:$LD_LIBRARY_PATH
MD 自动化处理
- # v100 /home/databank/zzy/project/D3PM/batch3/MD_auto_workdir
- # 该服务器conda 换成了miniforge,因此原来的一些包没了,我在zzy环境里配齐了
- mamba activate zzy
- export LD_LIBRARY_PATH=/home/chpeng/software/miniforge3/envs/gcc_12/lib:$LD_LIBRARY_PATH
- source ~/zzy/gmx2024.4-gpu.sh
- source ~/xcy/env/amber24.sh
- export OMP_NUM_THREADS=10
- export CUDA_VISIBLE_DEVICES=0,1,2
- # apo
- for i in {1..6..2}; do cp -r ../clean_system/system${i} ./;cp -r ../mdp/ system${i};done
- # complex
- for i in {2..6..2};do cp -r ../clean_system/system${i} ./;cp -r ../mdp/ system${i}/;done
- python total_control-v2.py 0
- # 成功完成
- # npt3.gro 检查无误
生成tpr
- # 从 v100 上将MD 自动化得到的结果同步至 75.3
- 75.3 /home/data/MD_traj_dataset/D3PM/batch3/1um
- source ~/.gmx2024.sh
- nohup bash do.sh