
9.8 batch4
体系挑选
自batch3开始,为了快速从D3PM中寻找简单体系去跑起来,使用了更高效的挑选方式:
一次性下载大量PDB结构,检查结构里的配体,挑出没磷酸根的体系记录离子,记录二硫键的体系
蛋白检查 & 清洗
PDBfixer
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/protein_preparation
- # 将batch4的体系一起放入 list (4HTZ_A这样的形式即可)
- # Pdbfix 只要给定了--pdbid 参数,可以自己从pdb网站获取输入结构(实操中因为HTTP error无法实现)
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/protein_preparation/wget_pdb
- # wget 从PDB 网站中下载文件
- for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do wget "https://files.rcsb.org/download/${i}.pdb";done - # Pdbfix --add-residues 可以将氨基酸序列补齐,但是也会将两端补上,这往往会导致错误
- # 将不补链的结果存储在no_add-residues文件夹
- # 将补链的结果存储在add-residues文件夹
- for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do pdbfixer ../wget_pdb/${i}.pdb --ph=7.0 --keep-heterogens=none --add-atoms=all --add-residues --output=${i}-pdbfixer.pdb;done - for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do pdbfixer ../wget_pdb/${i}.pdb --ph=7.0 --keep-heterogens=none --add-atoms=all --output=${i}-pdbfixer.pdb;done
统一氨基酸序列
- # 撰写脚本 do.sh,统计两端补链前后的变化,N端的补链均已删除(已晶体结构为准)
- # 该脚本会取出D3PM中记录的链,因此小分子也删去了
- # C端的未作删除,变化情况记录在nohup.out中
- # 检查下来,N端也可以先不删,因为部分体系apo-complex的N端开头不一样
- # 本次是手动将apo-complex中N端少的那一个补齐了
- # 检查N端,C端,apo-complex 需要保证至少氨基酸序列一致
- for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do echo "########################## $i ##########################";head -n1 ${i}*;done - for i in
`awk -F '_' '{print $1}' ../list|tr '[:upper:]' '[:lower:]'`;do echo "########################## $i ##########################";tail -n1 ${i}*;done
pdb2pqr加氢
- # 75.5 /home/databank/zyzhou/project/d3pm/batch4/protein_preparation/pdb2pqr
- ############################## do.sh #########################################################
- for i in {1..18}
- do
- a=
`sed -n "${i}p" ../list|awk -F '_' '{print $1}'|tr '[:upper:]' '[:lower:]'` - b=
`sed -n "${i}p" ../list|awk -F '_' '{print $2}'` - echo "########################## ${a} chain ${b} ##########################"
- awk '$12!="H"{print}' ../pdbfixer/${a}_${b}-pdbfixer-clean.pdb >${a}_${b}-noH.pdb
- pdb2pqr30 ${a}_${b}-noH.pdb ${a}_${b}.pqr --ff AMBER --ffout AMBER --with-ph 7.0 --pdb-output ${a}_${b}-pdb2pqr.pdb
- rm ${a}_${b}-noH.pdb ${a}_${b}.pqr
- done
- ###############################################################################################
配体处理
- # 7个体系,直接去pdb页面上下载sdf文件 23 /home/databank_70t/zzy/g16/D3PM/batch4/raw_from_pdb
- 23 /home/databank_70t/zzy/g16/D3PM/batch4/epik
- # 2021 薛定谔ligprep epik 对其进行质子化
- bash do.sh
- 23 /home/databank_70t/zzy/g16/D3PM/batch4/test
- for i in {2..14..2}; do a=
`sed -n "${i}p" ../system-name`;mkdir -p $a;cp ../epik/${a}-lig.sdf ${a}/;done - # 检查是否有报错体系
- python check.py -i /home/databank_70t/zzy/g16/D3PM/batch4/test -t sdf
- # 均通过(无报错)
- 23 /home/databank_70t/zzy/g16/D3PM/batch4/g16_cal
- date | sed "s/^/D3PM system: seven g16 jobs submitted on /" >>nohup.out
- nohup python lig_parameter_cal.py -i /home/databank_70t/zzy/g16/D3PM/batch4/g16_cal -t sdf
- # 任务已提交
特殊体系的处理
9-15 尚未处理
4htz 体系中含有两个离子,待添加至pdb2pqr输出文件中
3t5g 的补链要检查,应当用apo叠合后的坐标对口袋旁的结构进行补齐
4hzt_4d08_pair20
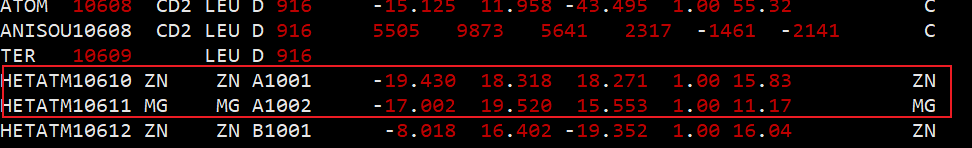
将PDB 中原始的离子坐标信息复制到pdb2pqr的结果中
注意,PDB文件中apo构象给出的信息是两个离子都不带电,而complex都是带电(zn2+)
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/protein_preparation/4hzt_4d08_pair20_ions
3t5g_pair26
将 4jvf的112-120坐标(晶体结构的112-117未解析出来)替换为3t5g 叠合到 4jvf后的112-120 坐标 (3t5g 的 M117与 4JVF的 M118 距离太远了,直到 A120 两个结构才叠合的较好,所以坐标替换截止到120)
mdframe
- 23 /home/databank_70t/zzy/g16/D3PM/batch4/test-zjhan
- nohup python -m mdframe --cpu 20 -t lig_param_run --config zzy.yaml -i /home/databank_70t/zzy/g16/D3PM/batch4/test-zjhan/g16_cal &
- # 对高斯优化模块做了测试,发现 sdf 电荷识别存在问题,等待师兄debug
- # 根据师兄提供的环境配置文件配置环境,即可完成mdframe的配置,脚本可从上下载后解压
- mamba env create -f mdfrmae_env.yaml -n mdframe
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/md_sys_pre/apo
- # 先测试mdframe对apo体系的准备情况,7个apo蛋白放置在 clean_protein_input_file文件夹
- for i in
`cat list`;do a=`echo $i|awk -F '_' '{print $1}'|tr '[:upper:]' '[:lower:]'`;mkdir $i;cp ../../protein_preparation/clean_protein_input_file/${a}* $i/;done
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/md_sys_pre/complex
- # 测试mdframe对complex体系的准备情况,7个apo蛋白放置在 clean_protein_input_file文件夹
- # 由于本次高斯优化是用的老版本准备的,所以命名要改一下,总的来说,输入的sdf文件命名里得包含'mol'
- for i in
`cat list`;do a=`echo $i|awk -F '_' '{print $1}'|tr '[:upper:]' '[:lower:]'`;mkdir $i;cp ../../protein_preparation/clean_protein_input_file/${a}* $i/;cp ../../ligprep/g16_cal/$i/${i}-lig.sdf $i/${i}-mol.sdf;cp -r ../../ligprep/g16_cal/$i/ligprep $i/ligprep_${i}-mol;mv $i/ligprep_${i}-mol/${i}-lig.log $i/ligprep_${i}-mol/${i}-mol.log;cp $i/${i}-mol.sdf $i/ligprep_${i}-mol/;done
生成tpr
- 75.5 /home/databank/zyzhou/project/d3pm/batch4/1us_cmd_tpr